

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 6, Number 2-3, September 2017, pages 62-67
De Novo Precursor B-Lymphoblastic Leukemia/Lymphoma With Double-Hit Gene Rearrangements (MYC/BCL-2) Presented With Spinal Cord Compression and Acquired Factor XIII Deficiency
Dina Sameh Solimana, b, e, Ahmad Al-Sabbagha, Feryal Ibrahima, Shehab Fareedc, Mohamed Talaatd, Mohamed A. Yassinc
aDepartment of Laboratory Medicine and Pathology, National Center for Cancer Care and Research, Hamad Medical Corporation, Doha, Qatar
bDepartment of Clinical Pathology, National Cancer Institute, Cairo University, Cairo, Egypt
cDepartment of Hematology and Medical Oncology, National Center for Cancer Care and Research, Hamad Medical Corporation, Doha, Qatar
dDepartment of Radiology, National Center for Cancer Care and Research, Hamad Medical Corporation, Doha, Qatar
eCorresponding Author: Dina SamehSoliman, Department of Laboratory Medicine and Pathology, National Center for Cancer Care and Research, Hamad Medical Corporation, Doha, Qatar
Manuscript submitted June 7, 2017, accepted June 29, 2017
Short title: De Novo Precursor B-Lymphoblastic Leukemia
doi: https://doi.org/10.14740/jh329w
| Abstract | ▴Top |
Double-hit lymphomas (DHLs) are aggressive mature B-cell neoplasms associated with rearrangements involving MYC and B-cell lymphoma-2 (BCL-2). Such DH events are extremely rare in B-cell precursor acute lymphoblastic leukemia (B-ALL), especially in young adults. A 29-year-old male patient initially presented to emergency department with right mandibular mass of 2 months duration associated with intermittent fever. Laboratory workup revealed very high lactate dehydrogenase at 2,026.0 U/L. Peripheral blood revealed pancytopenia with many circulating blasts (about 77%). Bone marrow (BM) aspirate revealed infiltration with many small sized blasts of very high nucleocytoplasmic ratio, finely dispersed nuclear chromatin and prominent nucleoli. The BM biopsy reflected marked hypercellularity with diffuse replacement by sheets of blasts, positive for TdT, PAX-5, CD10, cMYC, BCL-2 and CD20 with Ki-67 > 90%. Flow cytometry on BM revealed a precursor B-immunophenotype (CD45 (dim), CD19, CD10, Tdt and CD20). The blasts are negative for cytoplasmic and surface IgM. Cytogenetics revealed complex karyotype: 46,XY,del(6)(q21q23),t(8;22)(q24.1;q11.2),t(14;18)(q32;q21)(20). A diagnosis of B-lymphoblastic leukemia/lymphoma with t(8;22)(q24.1;q11.2) and t(14;18)(q32;q21) was made. Fluorescent in situ hybridization (FISH) analysis revealed an abnormal hybridization signal pattern for CDKN2A probe, indicating biallelic (homozygous) deletion of the short arm of chromosome 9 (9p) in 94% of the cells analyzed. The patient had severe life-threatening bleeding despite of normal prothrombin time (PT) and activated partial thromboplastin time (APTT) due to acquired factor XIII deficiency, an overlooked rare coagulopathy disorder. In addition, the patient developed acute sudden onset paraplegia, and magnetic resonance imaging (MRI) of spine showed acute cord compression which necessitated emergency radiotherapy after which chemotherapy was started on hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, adriamycin, and dexamethasone) protocol. MRI showed dramatic resolution of the mass. Very few cases of B-ALL with DH rearrangement with true precursor B-cell phenotype (positivity for TdT with negativity for surface light chain) have been reported. Many of these had frequent central nervous system (CNS) involvement, with complex karyotypes, highly aggressive course, with short survival of less than 1 year. This case however showed very good response to treatment. In contrary to DHL, de novo B-ALL with double-hit rearrangements is more prevalent in pediatrics and young adults. Although most of reported cases represent transformation of follicular lymphoma, our patient’s young age, acute onset and absent lymphadenopathies all support de novo ALL.
Keywords: Double hit B-ALL; MYC; BCL-2; Acquired FXIII deficiency
| Introduction | ▴Top |
Double-hit lymphomas (DHLs) are aggressive subcategory of large B-cell lymphomas (BCLs) that have MYC and BCL-2 and/or BCL-6 (“triple-hit” (THL)) rearrangements identified based on fluorescent in situ hybridization (FISH) or standard cytogenetics and are currently classified according to recent World Health Organization (WHO) classification (2016) as “high-grade B-cell lymphoma, with MYC and BCL2 and/or BCL6 rearrangements”, including all “double-/triple-hit” lymphomas other than follicular lymphomas (FL) or lymphoblastic lymphomas (B-LBLs) [1]. MYC/BCL2 DHL is a rare entity; the incidence of genetic DHL varies among different studies, ranging from 3% to 13% of diffuse large B-cell lymphoma (DLBCL) [2]. These neoplasms usually have a mature germinal center B-cell immunophenotype, a complex karyotype and an extremely aggressive clinical course with poor response to therapy [3, 4]. According to WHO classification (2008), most cases with MYC/BCL2 DH rearrangements have histologic features of DLBCL or BCL, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma (BCLU). In a very few occasions, DHL has the morphology and immunophenotype of B-LBL or FL [5].
| Case Report | ▴Top |
A 29-year-old male with no significant past medical history initially presented to emergency department with right cheek swelling of 2 months duration associated with intermittent fever, night sweats, dyspnea on exertion, and no significant weight loss. On examination, he was pale, with palpable mass in the right mandible 5 × 3 cm. No lymphadenopathies and no previous history of lymphomas were reported.
Biochemical workup showed normal urea, creatinine, uric acid, normal total bilirubin and liver function test with very high lactate dehydrogenase (LDH) 2,026.0 U/L (125 - 225 U/L).
His initial complete blood count (CBC) showed pancytopenia with absolute neutrophil count (ANC) at 0.1 × 103/µL, hemoglobin of 7.9 g/dL and platelet count of 59 × 103/µL. Peripheral smear showed pancytopenia with many circulating blasts (77%) which warranted bone marrow (BM) examination.
BM aspirate smear showed extensive infiltration by 97% blasts with markedly suppressed erythropoiesis and granulopoiesis. The leukemic cells were small in size with very high nucleocytoplasmic ratio and multiple prominent nucleoli. Few blasts showed fine cytoplasmic blebs/pseudopods (Fig. 1).
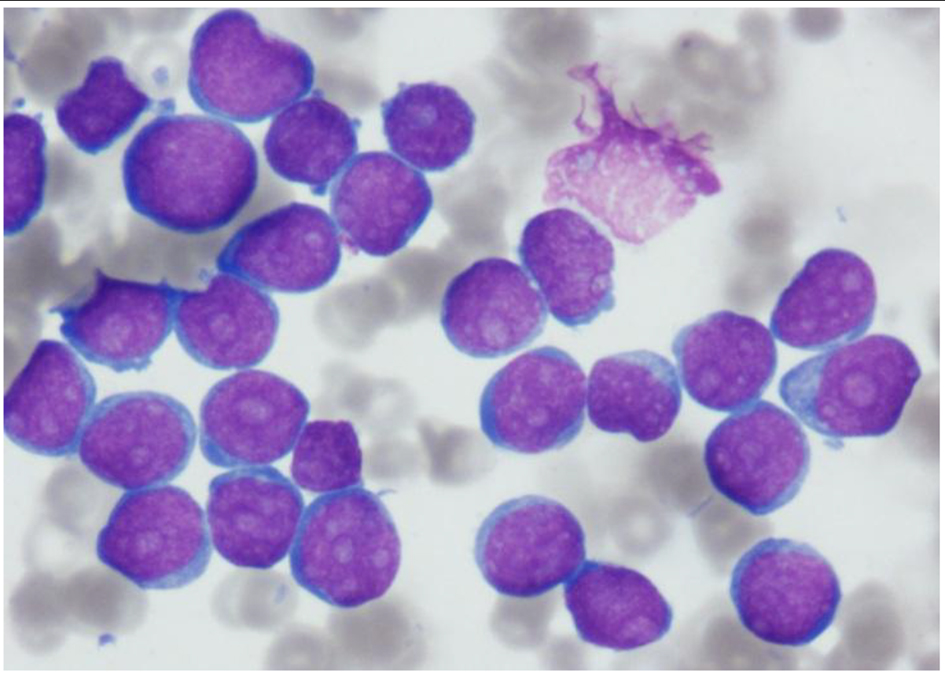 Click for large image | Figure 1. Aspirate smear showing extensive infiltration with blasts, small in size with very high nucleocytoplasmic ratio and multiple prominent nucleoli. Few blasts show fine cytoplasmic blebs/pseudopods (Wright stain, × 1,000 magnification). |
The BM biopsy reflected marked hypercellularity (about 100%), diffusely infiltrated with monomorphic population of small to medium sized lymphoid blasts with finely dispersed nuclear chromatin and prominent nucleoli (Fig. 2a). There was marked megakaryocytic proliferation noted, with some clustering (loose and dense) (Fig. 2b). By immunohistochemistry, the blasts were positive for TdT, PAX-5 (Fig. 3a, b), CD10 and CD20 with high proliferative index reflected by high Ki-67 (> 90% of the infiltration).
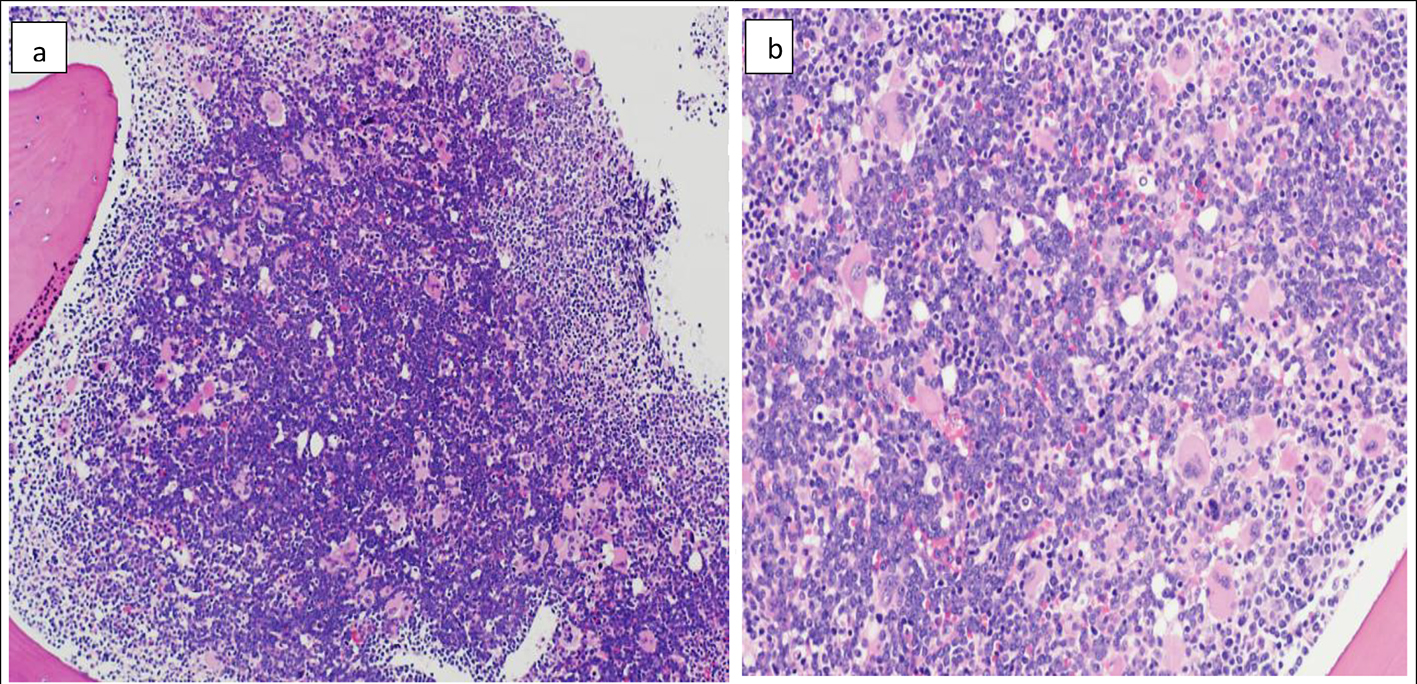 Click for large image | Figure 2. Bone marrow biopsy (H&E) showing marked hypercellularity (about 100%) and diffuse infiltration with monomorphic population of small to medium sized lymphoid blasts. The blasts in the center of the biopsy appear larger with more prominent nucleoli compared to periphery (a) (× 10). Marked megakaryocytic hyperplasia (b) (× 20). |
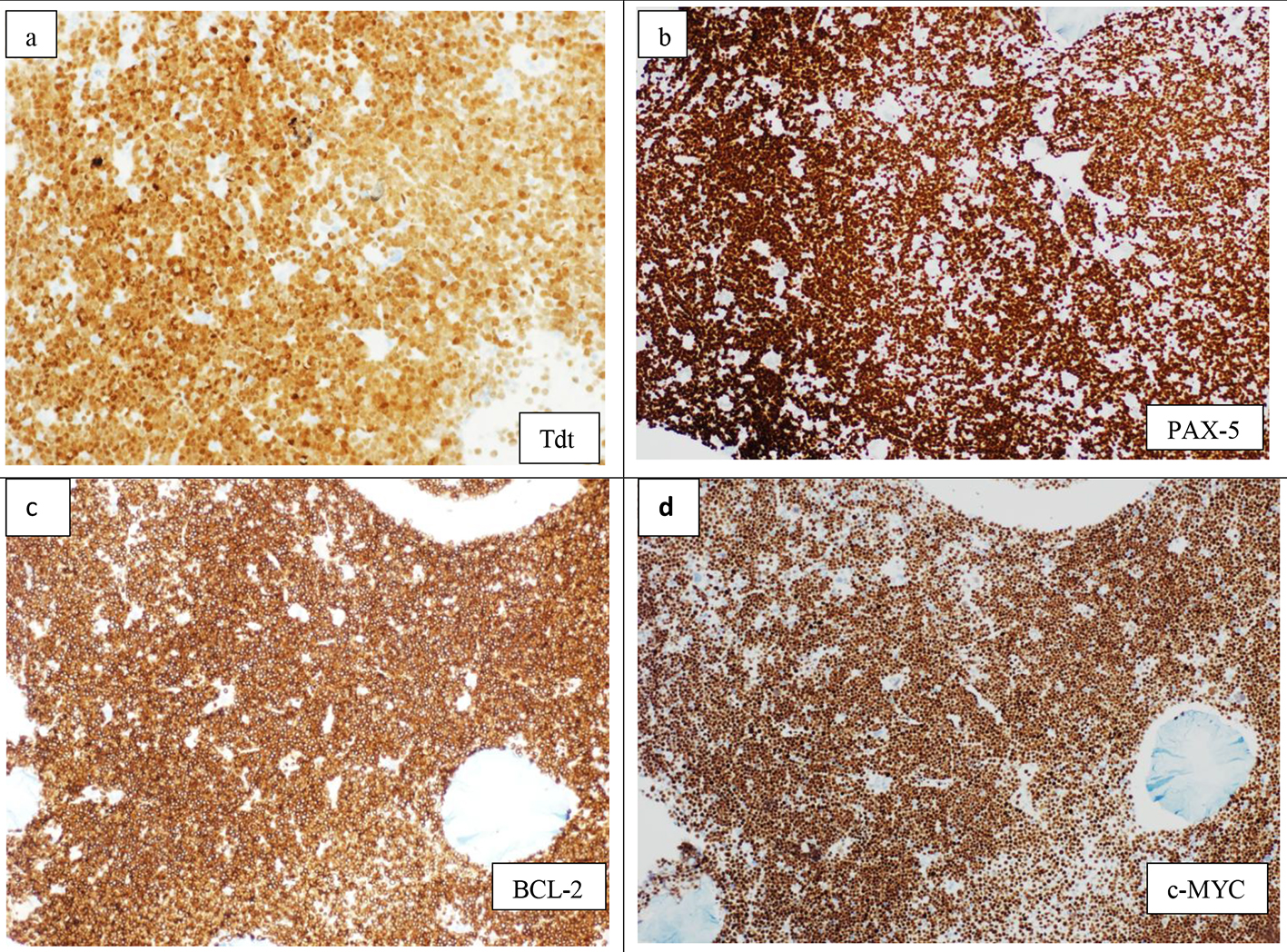 Click for large image | Figure 3. Immunohistochemistry on bone marrow biopsy showing diffuse infiltration by lymphoid blasts, positive for TdT (× 20) (a), PAX-5 (× 10) (b), BCL-2 (×10) (c), and c-MYC (× 10) (d). |
Flow cytometry on BM aspirate revealed 96% blasts with immunophenotype classic of precursor B-lymphoblastic leukemia (B-ALL): CD45 (dim), CD19, CD79a, CD10, HLADR, Tdt and CD20 with no significant expression of cytoplasmic IgM.
Cytogenetics study showed abnormal clone with deletion of the long of chromosome 6, a translocation between long arms of chromosome 14 and 18, t(14;18)(q32;q21), and a translocation between long arms of 8 and 22, t(8;22)(q24;q11.2).
Karyotype was 46,XY,del(6)(q21q23),t(8;22)(q24.1;q11.2),t(14;18)(q32;q21)(20).
FISH analysis revealed biallelic (homozygous) deletion of the short arm of chromosome 9 (9p) in 94% of the cells analyzed. cMYC, BCL-2 immunostains on BM biopsy showed strong diffuse positivity (Fig. 3c, d) with negative BCL-6. A diagnosis of B-ALL with combined t(8;22)(q24.1;q11.2) and t(14;18)(q32;q21) was made.
Few days later, the patient developed fever and massive pleural effusion was confirmed. Flow cytometry on pleural fluid showed malignant effusion with 96% of blasts of same phenotype described earlier.
Chest tube was inserted and debulking chemotherapy was started on steroids 50 mg, rituximab 375 mg/m2, cyclophosphamide 300 mg/m2, vincristine 2 mg (R-COP), and tumor lysis measures including hydration and rasburicase. The patient demonstrated evident bleeding tendency in the form of significant massive bleeding from BM aspiration site, massive hemothorax, and psoas muscle hematoma which necessitated detailed coagulopathy workup.
Routine coagulation studies revealed normal prothrombin time (PT): 14.0 s (9.4 - 12.5 s), international normalized ratio (INR): 1.3, partial thromboplastin time (PTT): 31.3 s (25.1 - 36.5 s) with high D-dimer at 4.51 mg/L FEU. Further coagulation factors assay was performed to unreveal the cause of severe bleeding tendency despite of normal PT and activated PTT (APTT) and revealed factor XIII deficiency at 38% (70-140%).
The patient then developed an acute onset paraplegia (power 0 out of 5) and sensory level up to T10. An urgent magnetic resonance imaging (MRI) of spine showed acute cord compression secondary to huge paravertebral and intraspinal mass in the lower dorsal and lumbar regions (Fig. 4a, b). An emergency radiotherapy to the spinal cord (10 fractions) was given.
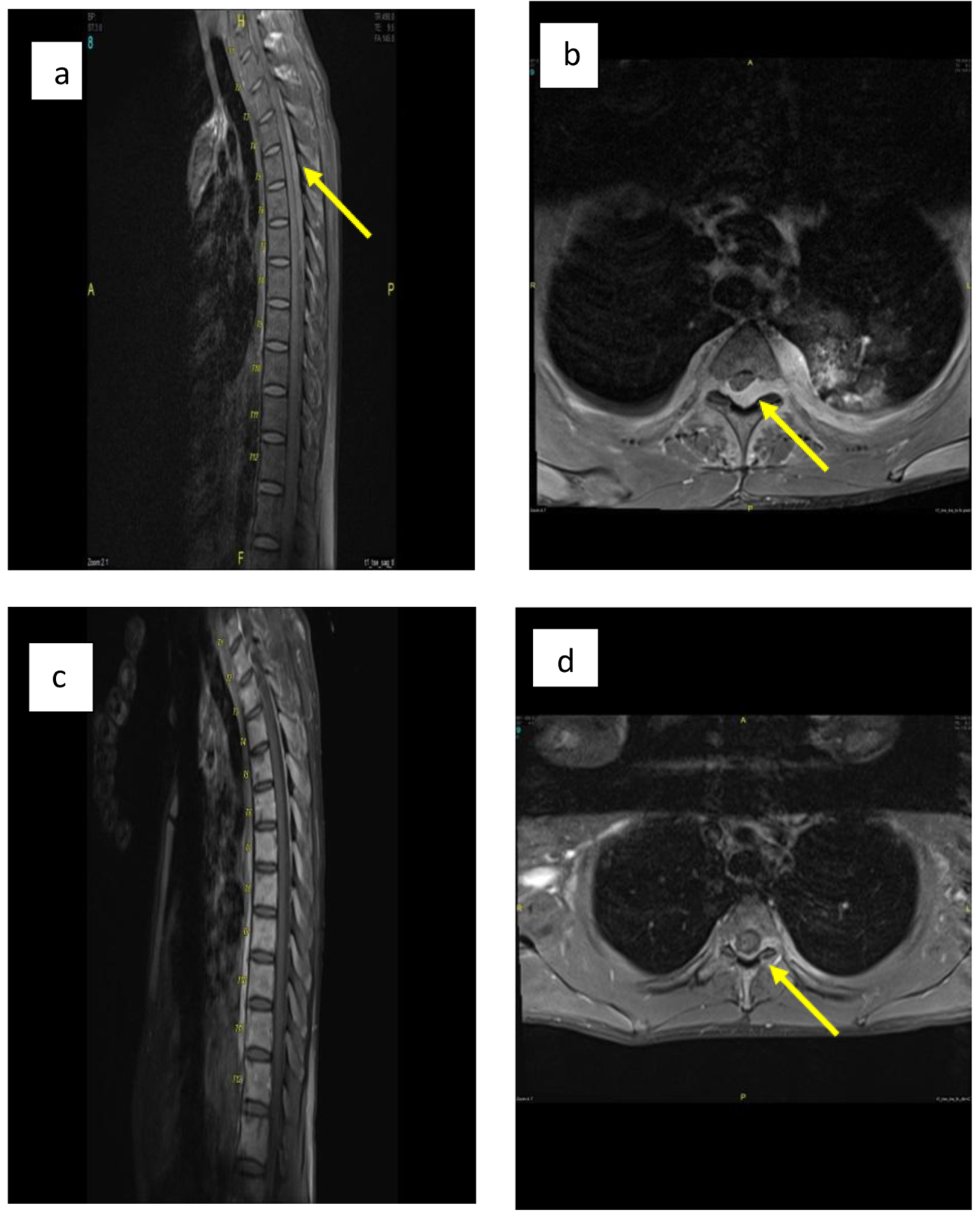 Click for large image | Figure 4. (a, b) Sagittal and axial post-contrast fat saturated images of thoracolumbar spine before treatment show posterior intraspinal extradural mass lesion extending from T2 to T8 levels in sagital image. Mildly compression are seen at the posterior aspect of cord, more from left side at T3 in axial image (arrow in b) with extension into both neural foramina more in left side as well as small paraspinal soft tissue component. (c, d) Sagittal and axial post-contrast fat saturated images at the same level after treatment show significant improvement with only small residual component seen posterior to cord at T3 level and extending into left neural foramina (arrow in d). |
Two weeks after radiotherapy, the patient was started on hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, adriamycin, and dexamethasone) protocol.
The patient completed three cycles of hyper-CVAD. After first cycle, the patient demonstrated a very good response with BM morphologic remission.
After completion of radiotherapy and chemotherapy, the patient improved dramatically with power reached 4/5 in his lower limb (with daily physiotherapy) and achieved complete remission confirmed by MRI of spine (Fig. 4c, d), positron emission tomography (PET) scan, CT and BM examination. The patient was in complete remission (CR) for 15 months after the initial presentation at last follow-up.
| Discussion | ▴Top |
Very few cases of MYC/BCL2 double-hit de novo ALL (DH de novo B-ALL) with true immature phenotype have been reported in English literature so far. Therefore, the clinical characteristics of this group of patients are not well characterized.
It was difficult to recognize the true incidence of cases of de novo acute B-ALL with MYC and BCL-2 rearrangements, and the reported cases so far are probably still overestimated as most of documented cases within this category either represent high-grade B-cell neoplasm with DH rearrangements and blastoid morphology but with mature phenotype [6, 7] which are currently classified as high-grade B-cell neoplasm with MYC and BCL-2 gene rearrangements [1] or cases of Burkitt leukemia/lymphoma (with L3 morphology [8]) or cases with blastic transformation on top FL [9, 10].
Cases with a well-documented true immature immunophenotype, positive for TdT and negative for surface light chains, are rare [4-10].
The largest relevant study performed by Liu et al [4], who screened 1,624 de novo B-ALL patients for concurrent 8q24 (MYC) and 18q21 (BCL2) translocations, and found only three patients who had an immature B immunophenotype with complex karyotype involving MYC and BCL-2 rearrangements.
Although Stamatoullas et al [11] reported five cases as B-ALL with both BCL-2 and MYC rearrangements, all the described cases showed mature phenotype and actually represented DHLs rather than precursor B-ALL.
Most patients with DHL are adults, often within elderly age group (with median age ranging from 51 to 65 years) and extremely rare in children and young adults [3, 4]. However, it seems that this is not the case for DH de novo B-ALL, as upon literature review, we found that true DH de novo B-ALL is more frequent among pediatrics and young adults similar to our case with age ranging between 15 and 24 years [4, 8].
Transformation of FL although rarely reported is a well-documented event [12, 13]. On the other hand, transformation of DLBCL (GC) into B-ALL is a very rare event with a single case reported [9]. Our team has also recently reported a case of Tdt positive high-grade B-cell neoplasm evolved in a case of DLBCL (ESH 2017poster presentation [14]).
Although our case showed classic immunophenotypic and morphologic characteristics of B-ALL, the aggressive presentation with extramedullary pattern of involvement raised the suspicion of lymphoma rather than acute leukemia. This was also reinforced by detection of DH rearrangements. The possibility of lymphoblastic transformation on top of FL in this case was confounded, given the patient young age with acute sudden onset and the classic precursor B-ALL morphologic and immunophenotypic profile with diffuse BM involvement and absent paratrabecular localization that is characteristic of FL.
In addition, careful clinical history and examination failed to detect any clinical, radiological or pathological evidence of significant lymphadenopathies/masses or history of previous underlying lymphoma.
Cases of DH de novo B-ALL described before share many similarities with DHL and that of transformed FL, including very poor prognosis, with all reported survival durations of less than 1 year. In the same way, our case shared many of the previously reported clinical characteristics. However, in contrast to most other reported cases, this case achieved a very good response to hyper-CVAD.
A single reported case of a 15-year-old patient [4] had also demonstrated similar good response with intensified treatment regimen under COG protocols and was in CR 15 months after initial diagnosis.
In addition, this case suffered severe life-threatening bleeding despite of normal PT and APTT. The bleeding was far beyond what is usually seen in leukemia patients and necessitated more extensive coagulopathy workup which pointed to acquired factor XIII deficiency, an overlooked rare coagulopathy disorder.
Although publications about cancer-associated factor XIII date back to the 1970s, and although factor XIII can be easily and safely substituted, only limited data on acquired factor XIII deficiency in cancer patients are available so far [15-17].
First-line treatment for cancer-associated coagulopathy including life-threatening hyperfibrinolysis and disseminated intravascular coagulation (DIC) is the treatment of the malignancy itself [18].
Another morphologically interesting feature in this case is the presence of marked megakaryocytic hyperplasia (Fig. 2b), a finding rarely seen in acute leukemia in which the BM is usually replaced by sheets of blasts suppressing all marrow elements. The presence of an underlying/coexisting myeloproliferative neoplasm was soon excluded by molecular studies for JAK2 and CALR mutations analysis that came to be negative. Since the patient had acquired factor XIII deficiency, it was suggested that megakaryocytic proliferation could be a compensatory reactive mechanism to patient’s active bleeding. This megakaryocytic hyperplasia was not detected in subsequent follow-up BM biopsies which were also correlated with normalized factor XIII level.
Conclusion
Very few cases of B-ALL with DH rearrangement with true precursor B-cell phenotype (positivity for TdT with negativity for surface light chain) have been reported. Extensive or unusual pattern of involvement in this case despite the classic morphologic and immunophenotypic profile of precursor B-ALL had prompted suspicion and urged further workup especially screening for DH gene rearrangements by FISH.
It is of clinical and diagnostic importance to recognize and characterize this rare entity and to identify its molecular pathogenesis as these aggressive lymphoid neoplasms may necessitate a customized and perhaps more intensive treatment protocols. Large series studies are needed to highlight more of disease characteristics including disease prognosis and response to therapy.
| References | ▴Top |
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, et al. The 2016 revision of the World Health Organization (WHO) classification of lymphoid neoplasms. Blood 2016;127(20):2375-2390.
pubmed doi - Petrich AM, Nabhan C, Smith SM. MYC-associated and double-hit lymphomas: a review of pathobiology, prognosis, and therapeutic approaches. Cancer. 2014;120(24):3884-3895.
doi pubmed - Nacheva E, Dyer MJ, Fischer P, Stranks G, Heward JM, Marcus RE, Grace C, et al. C-MYC translocations in de novo B-cell lineage acute leukemias with t(14;18)(cell lines Karpas 231 and 353). Blood. 1993;82(1):231-240.
pubmed - Liu W, Hu S, Konopleva M, Khoury JD, Kalhor N, Tang G, Bueso-Ramos CE, et al. De Novo MYC and BCL2 Double-hit B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) in pediatric and young adult patients associated with poor prognosis. Pediatr Hematol Oncol. 2015;32(8):535-547.
doi pubmed - Li S, Lin P, Fayad LE, Lennon PA, Miranda RN, Yin CC, Lin E, et al. B-cell lymphomas with MYC/8q24 rearrangements and IGH@BCL2/t(14;18)(q32;q21): an aggressive disease with heterogeneous histology, germinal center B-cell immunophenotype and poor outcome. Mod Pathol. 2012;25(1):145-156.
doi pubmed - Mufti GJ, Hamblin TJ, Oscier DG, Johnson S. Common ALL with pre-B-cell features showing (8;14) and (14;18) chromosome translocations. Blood. 1983;62(5):1142-1146.
pubmed - Chapiro E, Radford-Weiss I, Cung HA, Dastugue N, Nadal N, Taviaux S, Barin C, et al. Chromosomal translocations involving the IGH@ locus in B-cell precursor acute lymphoblastic leukemia: 29 new cases and a review of the literature. Cancer Genet. 2013;206(5):162-173.
doi pubmed - Pegoraro L, Palumbo A, Erikson J, Falda M, Giovanazzo B, Emanuel BS, Rovera G, et al. A 14;18 and an 8;14 chromosome translocation in a cell line derived from an acute B-cell leukemia. Proc Natl Acad Sci U S A. 1984;81(22):7166-7170.
doi pubmed - Loghavi S, Kutok JL, Jorgensen JL. B-acute lymphoblastic leukemia/lymphoblastic lymphoma. Am J Clin Pathol. 2015;144(3):393-410.
doi pubmed - D'Achille P, Seymour JF, Campbell LJ. Translocation (14;18)(q32;q21) in acute lymphoblastic leukemia: a study of 12 cases and review of the literature. Cancer Genet Cytogenet. 2006;171(1):52-56.
doi pubmed - Stamatoullas A, Buchonnet G, Lepretre S, Lenain P, Lenormand B, Duval C, Callat MP, et al. De novo acute B cell leukemia/lymphoma with t(14;18). Leukemia. 2000;14(11):1960-1966.
doi pubmed - De Jong D, Voetdijk BM, Beverstock GC, van Ommen GJ, Willemze R, Kluin PM. Activation of the c-myc oncogene in a precursor-B-cell blast crisis of follicular lymphoma, presenting as composite lymphoma. N Engl J Med. 1988;318(21):1373-1378.
doi pubmed - Fiedler W, Weh HJ, Zeller W, Fonatsch C, Hillion J, Larsen C, Wormann B, et al. Translocation (14; 18) and (8; 22) in three patients with acute leukemia/lymphoma following centrocytic/centroblastic non-Hodgkin's lymphoma. Ann Hematol. 1991;63(5):282-287.
doi pubmed - Training course WHO classification: towards personalized medicine in hematology. Saggart (Dublin), Ireland 9-11 March, 2017.
- Rasche H, Haghou F, Gaus W, Dietrich M, Hoelzer D, Pflieger H, Kurrle E, et al. [Blood clotting factor XIII substitution in acute leukaemia: result of a randomized and controlled study]. Dtsch Med Wochenschr. 1982;107(49):1882-1886.
doi pubmed - Sutor AH, Mall V, Thomas KB. Bleeding and thrombosis in children with acute lymphoblastic leukaemia, treated according to the ALL-BFM-90 protocol. Klin Padiatr. 1999;211(4):201-204.
doi pubmed - Weltermann A, Pabinger I, Geissler K, Jager U, Gisslinger H, Knobl P, Eichinger S, et al. Hypofibrinogenemia in non-M3 acute myeloid leukemia. Incidence, clinical and laboratory characteristics and prognosis. Leukemia. 1998;12(8):1182-1186.
doi pubmed - Fiegl M, Weltermann A, Stindl R, Fonatsch C, Lechner K, Gisslinger H. Massive disseminated intravascular coagulation and hyperfibrinolysis in alveolar rhabdomyosarcoma: case report and review of the literature. Ann Hematol. 1999;78(7):335-338.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.