

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Review
Volume 1, Number 2-3, June 2012, pages 44-53
Thrombocytopenia in Adults: Review Article
Mehmet Ali Erkurta, b, Emin Kayaa, Ilhami Berbera, Mustafa Koroglua, Irfan Kukua
aDepartment of Hematology, Faculty of Medicine, Inonu University, Malatya, Turkey
bCorresponding author: Mehmet Ali Erkurt, Department of Hematology, School of Medicine, Turgut Ozal Medical Center, Inonu University, TR-44280 Malatya, Turkey
Manuscript accepted for publication June 12, 2012
Short title: Thrombocytopenia in Adults
doi: https://doi.org/10.4021/jh28w
- Abstract
- Definition
- Diagnosis
- The Physiopathological Classification of Thrombocytopenias
- Common Etiologies
- References
| Abstract | ▴Top |
Thrombocytopenia is the result of falling the number of platelet from 150,000/microL. There are three main reasons of thrombocytopenia, a-Decreasing of making platelet b-Increasing of destruction platelet c-Changing of distribution platelet. Pseudothrombocytopenia must be kept in mind too. Both hereditary and acquired reasons help thrombocytopenia have wide spreaded, but acquired causes are more common with increasing age. Thrombocytopenia separates three stages as numerical. Mild: 100,000 - 150,000/microL, Moderate: 50,000 - 100,000/microL. Severe: < 50,000/microL. However, thrombocytopenia is not usually detected clinically until the platelet count has fallen to levels below 100,000/microL. Severe thrombocytopenia, such as intracerebral and intra-abdominal bleeding may be life threatening. So diagnosing the treatment immediately can save the life. Transfusion of platelet may not need in all thrombocytopenias. Treatment of the underlying disease may be sufficient. The reason of thrombocytopenia can be temporary but also can be caused severe diseases. Causes of thrombocytopenia change development levels of countries, according to geographical distribution and application centers. In this review we emphasize common etiologies seen in adult patients with thrombocytopenia.
Keywords: Thrombocytopenia; Etiology; Platelets; Treatment
| Definition | ▴Top |
Platelets were diagnosed the first time in 1860 by Zimmerman. The role of clotting blood was suggested in 1878 by Zimmerman and Hayran. They are the shape of disc which are 2 - 4 µm, 0.5 µm wideness, colorless, seedless. The average volume is 6 - 10 fL. There are about 150,000 - 450,000/microL platelet in pheripheral circulation of blood. The average life of them is 8 - 10 days. Nearls 2/3 platelet is in blood and 1/3 is in spleen [1]. Platelets can have a role both in primer and in seconder hemostasis. Platelets are essential for maintaining the integrity of the vascular endothelium and controlling hemorrhage from small-vessel injury through the formation of platelet plugs. More extensive injury and involvement of larger blood vessels requires, in addition to platelets, the participation of the coagulation system to provide a firm, stable, fibrin clot [2].
The average of the platelet counts should be 150,000 - 450,000/microL. If the numbers go down 150,000/microL, it causes thrombocytopenia. 2.5 percent of the normal population have got thrombocytopenia. There can be bleeding after mild trauma at patients with thrombocytopenia. The deficiency of platelets or functional out of order causes some symptoms. In general, if the number of platelets is over 100,000/microL, it can’t be expected bleeding even have an operation. It may last longer than usual bleeding between 50,000 - 100,000/microL in serious injuries. Even there can be bleeding between 20,000 - 50,000/microL at mild trauma. There can be bleeding itself the lower numbers such as 20,000/microL. There can be severe risks under 10,000/microL [3].
| Diagnosis | ▴Top |
Thrombocytopenia is not a disease but is a diagnosis. The detailed knowledge must be acquired from patients who have been suffering from the thrombocytopenia. Detailed examining and the test of laboratory should be done which are related to etiology. Some of the situation about thrombocytopenia must be checked. Recent new drugs or drugs that are only taken intermittently, recent infection, previously diagnosed hematologic disease, nonhematologic diseases known to decrease platelet counts (eg, eclampsia, sepsis, DIC, anaphylactic shock, hypothermia, massive transfusions), positive family history of bleeding and/or thrombocytopenia, recent live virus vaccination, poor nutritional status, pregnancy, recent organ transplantation from a donor sensitized to platelet alloantigens, recent transfusion of a platelet-containing product in an allosensitized recipient. History pertaining to alcohol consumption and HIV risk factors should be obtained. When faced with an asymptomatic patient with a low platelet count, the clinician should initially seek to exclude artifactual or “pseudothrombocytopenia” as the etiology. Also the story of the family must be questioned for the congenital thrombocytopenia reasons [4]. Thrombocytopenia is not only bleeding for patient, but it depends on heparin. Dissemine intravaskuler coagulation and as paroksismal nocturnal hemoglobinuria can be with thrombosis clinic. In physical examination the thrombocytopenia which depends on diagnosis petechia, purpura, nose bleeding, gum bleeding, hematuria, menorrhagia or as cerebral hemorrhage is understood also some symptoms which depend on thrombosis can be fixed. The peripheric smear must be done every patient who is thrombocytopenia. In normal magnify there can be seen 3 - 10 platelet in every part. With peripheric smear pseudothrombocytopenia can be eliminated in patient or the abnormality which causes thrombocytopenia can be recognized [5].
| The Physiopathological Classification of Thrombocytopenias | ▴Top |
There are three major patophsiological mechanisms in thrombocytopenia: reduced producing, rapid demolishing and sequestration. Rapid demolishing is the reason thrombocytopenia which is often seen. There has been existed when the rate of demolishing platelet passed the rate of producing. Demolishing of platelet depends on intracorpuscular or extracorpuscular reasons. Not only as sendrom of Wiscot-Aldrich but in some of the hereditary disease also can be seen thrombocytopenia which depends on intracorpuscular defects. Thrombocytopenia which depends on deficiency of producing myelosupressive drugs, radiation and as aplastic anemia can be seen due to suppression of megakaryocyte in bone marrow. Pure megakaryocytic hypoplasia or aplasia is a rarely situation. Amegakaryocytic thrombocytopenia, macrocytosis or like diserythropoiesis, with other disorder of cell series come together. It can be congenital or acquired. Due to capturing of platelets which are enlarging in spleen there can be seen secondary thrombocytopenia distinguishing of abnormal platelet. Sepsis, preeclampsia and immun thrombocytopenia in these panorama especially high rate of ruining platelet, the average quantity of platelet is high, disease such as aplastic anemia in which producing platelet is becoming less [6-8]. Physiopathological classifying of thrombocytopenia can be seen in detailed Table 1.
 Click to view | Table 1. Classification of Thrombocytopenia |
| Common Etiologies | ▴Top |
Artificial thrombocytopenia
Sometimes the platelet counts can be less even though it may not be thrombocytopenia. The persons who have got thrombocytopenia but also they don’t have petechia and ecchymosis must suspect about artificial thrombocytopenia. This situation accurs due to giant thrombocyte and thrombocyte satellitism, the most important common is pseudothrombocytopenia. Formation of thrombocyte in pseudothrombocytopenia occurs anticoagulin which depends on platelet agglutinant. The most farming occurs in EDTA when it is used. It occurs in anticoagulant when they come together such as citrate, acid citrate, dextrose, oxalate and heparin. In most of the patients forming activity of platelets increases under 37 °C, 22 °C or 4 °C is maximum level. Forming the platelets accurs in several minutes and it increases 60 - 90 minutes. They are called platelet cold agglutinate. Platelets in vitro condition which blood with anticoagulant on the epitope (usually GPIIb/IIIa complexity knowing antibody can form IgG). EDTA separates calcium from GPIIb/IIIa complexity and it causes new epitopes on GPIIb. Antibodies sometimes both platelet GPIIb/IIIa with epitope and also entering the reaction with neutrophil and leukocyte FcyIII receptor and it helps the platelets to stick to the round of leucocytes as a rosette. It is called platelet satellitism. The giant platelets are counted in blood measuring machine as it supposed lymphocyte; in fact they cause the low platelet counts [1].
Immune thrombocytopenia
Immune thrombocytopenia is an autoimmun illness which is developed against platelets auto antibodies and owing to destruction of premature platelets. The number of platelet in ITP is under the level 150,000/microL. The lifetime of platelet cut down. There are antiplatelet antibodies in plasma. Although most of ITP are autoimmune especially viral infection starts this. Antibody of antiplatelet lgG, stick to the membran of palelelets are kept through the macrophage Fc receptors in spleen, causes levaning from circulation. So platelet counts in circulation decreases [5]. In scintigraphy works the lifetime of platelet decreases from 5 - 7 days to 1 - 4 hours [9].
Although ITP can be seen in every age especially, it can be more seen at children and young. The rate in women is one and half times more often than men. The acute ITP which is seen in childhood shows acute bleeding. Chronic ITP which is shown in teenagers starts insidiously. Bleeding is spontaneously, it increases after trauma and bleeding which are different parts of body can be seen at same time. The skin bleeding is like purpura and ecchymosis. Epistaxis, gum bleeding, vaginal and gastrointestinal bleeding can be seen. Hematuria, cerebral and retinal bleeding, menorrhagia can be seen in women. For the diagnosis of ITP medical history and with the completing physical examining in laboratory researching the reasons which are producing seconder thrombocytopenia should be eliminated [10, 11]. The most important laboratory finding is thrombocytopenia and platelet anisocytosis. There have been platelets which are big and small in the shape of atipic in widespreading of blood. If there is loosing blood at the beginning even normochromic normocytes finds anemia, after the long bleeding the deficiency of iron can be seen. The number of leukocyte is usually normal. If there is loosing blood, it can be seen leucocytosis. The Associate of American Hematology, do not suggest researching of bone marrow especially under the age of 60 and the one who responses well treatment [5]. With these International Consensus suggest myelodysplasia and over the age of 40. Bone marrow is normal. Megakaryocytes may be increased. Immature, megakaryocyte which is big and has got one nucleus can be seen [12].
Risk of bleeding is related platelet number’s of chronic ITP which is seen in adults. The platelet counts are over 30,000/microL it is followed without treatment. But if a great operation will be treatment is necessary. Although the number of platelet is under 30,000/microL or over 30,000/microL, and also if there is mucous bleeding, it should be treated. In ITP of mature first step is drug corticosteroid and 1 - 2 mg/kg/day should be given. The patients who responses treatment well the number of platelet raises in first week and later it raises between two and four weeks. If there is bleeding which is threat the life, they should have done parenteral corticosteroid or intravenous immunoglobulin [5, 10]. The 20-30% percent of patients can not give response to the corticosteroid. Although they have taken corticosteroid for six months and can not give response, the severe thrombocytopenia (lower than 10,000/microL ) continue or the patients who take treatment for three mounts and then platelet counts lower than 30,000/microL are suitable to take out spleen. After increasing the number of platelets and in order to keep the platelet at this level and the patients who need 15 mg and over it prednisolone everyday, in spite of patients who take low dose steroid, have got side effects of it and condition contraindicate of steroid are needed to splenectomy for the second step therapy. They can have complete remission 60-80% after splenectomy. The patient whose platelet counts increases after operation 24 - 48 hours is responded well to splenectomy. If there is not any response to splenectomy, you should find accessory of it. Patients who have not response to splenectomy, thrombopoetin agonist and rituximab are used in third step therapy. The other treatments are azathioprine (3 - 4 mg/kg/day), cyclophosphamide (2 mg/kg/day) 6-mercaptopurine (100 mg/m2/day), vinkristin sulphate are used 1 - 2 mg three-four weeks [10, 12].
Infectious
As Cytomegalovirus, Epstein-Barr virus and Hepatitis occur thrombocytopenia during viral infections. Mycoplasma, mycobacterium, malaria and many infectious diseases such as brucellosis can be seen in the course of thrombocytopenia. Thrombocytopenia in many infectious appears less production thrombocytopenia but sometimes it occurs immune mechanism. The most important reason for thrombocytopenia which is seen at patients with sepsis is thrombocytopenia phagocytosing which happens rising of M-CSF (Macrophage Colony Stimulating Factor). Thrombocytopenia is also common in HIV-infected patients [13].
Leukemia
Leukemia is a clonal illness which is defect rise of immature cells in lenfoid and myeloid series. Leukemia consists of two groups: a) Acute leukemia (myeloid, lymphoid) and b) Chronic leukemia. Especially in acute leukemia, 1/3 of patients have petechia, ecchymosis and nose bleeding relation with thrombocytopenia because of bone marrow infiltration [1].
Drug-induced thrombocytopenia
Drugs cause the acute and severe bleeding. Thrombocytopenia occurs in different mechanism. Although some drugs causes the bone marrow suppression or pressuring the megakaryocyte in producing thrombocytopenia. Some drugs directly affect platelet. In most of the drugs there has been thrombocytopenia with antibody in some of the sensitive people. The drugs which are used in cancer treatment and also caused bone marrow suppression are cytostatic. These drugs suppress bone marrow and cause pancytopenia. Busulfan, melphalan, etoposide cyclophosphamide, folic acid antagonists, antimitotics, cytostatic antibiotics can cause thrombocytopenia by suppressing megakaryocyte. Immunological thrombocytopenia occurs in ways that drugs quinine, quinidine, phenacetin, methicillin, penicillin, sulfonamides, gold salts, and drugs such as heparin. These drugs which accurs some of the antibodies with platelets compheman reason fixing. Result of this platelets turns into destruction and developed thrombocytopenia. The immun thrombocytopenia depends on drugs occurs in three mechanism, i) The type of kinidin: The antibody occurs when the drug is taken for the first time. This antibody, when the drug is taken second time, it takes out complexity with drug, this complexity which connecting platelet’s membrane causes platelets go away from the circulation; ii) The type of penicillin: The drug connects directly to the platelet membrane and it causes leaving the platelets rapidly from the circulation; iii) The type of heparin: Heparin creates a complex with platelet fact 4 (TF4) which is forming from the platelet’s alpha granule is a cationic protein. The antibodies which are developed against to this complex lgG connect complex heparin TF4 on platelet cell membrane, endothelia cell membrane in plasma and then cause releasing platelet, thrombosis and decreasing platelet counts [13]. The drugs which cause thrombocytopenia are shown in Table 2.
Click to view | Table 2. Drugs Commonly Associated With Thrombocytopenia |
Heparin-induced thrombocytopenia (HIT)
Heparin causes the light and temporary decreasing in the number of thrombocyte to ranging from developing serious thrombosis and DIC (Disseminated intravascular coagulation). Heparin in therapeutic concentration of plasma (0.1 - 0.4 U/mL) can directly connected to the platelet and causes thrombosis and thrombocytopenia. Unfractionated heparin which has got much more platelet affinities than low molecular weight heparin. Heparin TF4 complexity Fcylla receptor on the platelet’s surface activates platelets which is connecting cross. An immune complex which exists in circulation and consists of antibody-heparin-TF4 can responsible for developing thrombosis result of connecting heparin which connects TF4 on platelets stimulate of secretion and aggregation [14, 15]. HIT clinic occurs in two types. i) The type of non-immunology: Platelet counts can not go down under 50,000/microL, there is a mild thrombocytopenia. Giving high dose intravenous heparin shortly after it can be seen and the thrombocytopenia can be better during the treatment of heparin. It is possible that it is attacked to heparin platelets which affect direct agglutination. ii) The type of immunologic: It can be seen starting to treat of heparin after 5 - 8 days. If the patient has used the heparin before, it may occur in a short time. Severe thrombocytopenia can be seen. It can be both thrombosis. Bleeding is rarely seen, the main problem is thrombosis. Venous thrombosis is much more than arteriyel thrombosis. Thrombosis developing patients the mortality and morbidity is high [16]. For the treatment of non-immunologic type it is sufficient to cut down heparin. Treatment of immunologic type, danaparoid which is alternative anticoagulant as a treatment, recombinant hirudin, ancrod and argatroban can be used. The HIT developing patients after cutting down the heparin we can start warfarin in low starting dose. Giving warfarin in high dose causes the decreasing of protein C and this result in thrombosis. It can be plasmapheresis to the severe thrombocytopenia patients due to thrombosis [17].
Chronic liver disease
The reasons of thrombocytopenia is at chronic liver illness secondary to portal hypertension splenomegaly, as immunology demolishing of platelets and the third one is lessening of production thrombopoetin. In addition the viral agent such as hepatitis C virus (HCV) of myelosupressive activities and the toxic effect overdose alcohol [18] and also treatment in chronic hepatitis interferon helps thrombocytopenia [19].
Megaloblastic anemia
Thrombocytopenia was seen in 20% of patients with megaloblastic anemia due to vitamin B12 and folic acid deficiency. It is improved because of ineffective thrombocytopoiesis. Rarely, severe thrombocytopenia may occur. In chronic alcoholics, thrombocytopenia occur the result of the liver cirrhosis with congestive splenomegaly or deficiency of folic acid. In some cases thrombocytopenia may develop in a 5 - 10 day period by direct effect of the alcohol which is taken overdose on bone marrow even without nutritional deficiency and splenomegali too; and the number of platelet may rise normal degree in a 5 - 21 day period after the alcohol is cut off [20].
Thrombocytopenia in pregnancy
Asymptomatic thrombocytopenia can be seen 5% in normal pregnancy. The 15% of women who are preeclampsia can be seen severe thrombocytopenia. Thrombocytopenia which happens with pregnancy is called gestational thrombocytopenia. There are 5 criteria of gestational thrombocytopenia: 1-Mild and asymptomatic thrombocytopenia, 2-There hasn’t been the story of thrombocytopenia before (except the previous pregnancy), 3-It can be only seen late period of pregnancy, 4-There hasn’t been togetherness with fetal thrombocytopenia, 5-It can be better as spontaneous after pregnancy. The number of platelet in gestational thrombocytopenia is over 70,000/microL. The 2/3 of patients are between 130,000 - 150,000/microL. The reason of thrombocytopenia which can be seen in pregnancy is unknown. Since clinic and laboratory diagnosis are resemble to the mild ITP it is supposed be autoimmune. Thrombocytopenia of pregnancy does not change following period of mother Birth should be done normal obstetric rules [13].
Aplastic anemia
Aplastic anemia is a deficiency of stem cell which consist pancytopenia and aplasia in bone marrow. Beside the deficiency of stem cell, abnormality microenvironments of bone marrow, the system of immune dysregulation and decreasing of growth factors such as many factors contribute developing aplastic anemia. Bone marrow is hypocellular and the bone marrow has been filled fat and stromal cell. In aplastic anemia thrombocytopenia which depends on deficiency of bone marrow can be the first finding of disease which is unknown. Skin and mukoza bleeding can often be seen at clinic [21]. The patients are under 40 ages and who also severe disease and if suitable donor was found, at first treatment allogeneic bone marrow transplantation should be considered [22].
Thrombocytopenia due to splenic pooling
With working kinetics of platelet at patients who are splenomegalies in have shown that the 90% of body’s total platelet which are kept in enlarged spleen caused for thrombocytopenia. Platelet which is seen with hypersplenism in thrombocytopenia moves to enlarged spleen from the circulation, as reversible and temporary sticks to the macrofaj which are in spleen. Thrombocytopenia which is seen in hypersplenism is usually middle degree and clinical importance is developed after coagulation disorders. Hypersplenism can be often seen in chronic liver disease with portal hypertension and congestive splenomegaly. In homozygous sickle cell anemia (period of before the splenic atrophy), hemoglobin C disease and thalassemia major, chronic infections, gaucher disease, myelofibrosis and lymphoma, spleen can enlarge. Usually, it does not need treatment. If the splenectomy is done, the number of platelet turns to normalcy, thrombocytosis can occur.
Thrombotic thrombocytopenic purpura
Thrombotic Thrombocytopenic Purpura (TTP) is a syndrome which is consist of little veins with widespreading thrombotic occlusion characterized microangiopatic hemolytic anemia, thrombocytopenia, neurological symptoms, fever and disorders of kidney. Von Willebrand Factor (vWF) that is released from endothelial cell is the shape of big molecular multimers. vWF which is swelling into the plasma, ADAMTS 13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) which is a metalloproteas, is another name von Willebrand Factor cleaving protease (vWF CP) breaks into little pieces by its enzyme. In congenital TTP although there is producing error in enzyme, there are antibodies which are designed against this enzyme in acquired formation. Thrombosis is made of thrombocyte in terminal arteriole and capillary, vWF and a little fibrin. Microaneurisms occur in thrombosis area. Hyaline accumulates in subendothelial layer. Thromboses mostly are seen in brain, kidney, pancreas, heart, spleen and adrenal gland. Erythrocytes which are passing through the veins in trombosis breaks into pieces and causes microangiopatic hemolytic anemia In addition to this, thrombocytopenia and coagulopathy occur in trombosis due to the consumption of trombosis and clotting factors [23]. The most effective treatment of TTP is plasmapheresis. Patient should be controlled a long time with plasmapheresis is made at regular intervals. Corticosteroids can be useful in suppressing antibodies which are developed against protease breaking vWF. Antiagregans can prevent platelet aggregation and occurring thrombosis. Splenectomy can be practiced in patient of refractory TTP [24].
HELLP syndrome
Hellp syndrome which is characterized hemolysis, increasing the liver enzyme and thrombocytopenia is pregnancy complication. The preeclampsia of 0.1-0.6% in all pregnancy and in eclamptic patients of 3-19.3% can be occurred [25]. Intravascular platelet activation occurs result of the damage of microvascular endothelium. Thromboxane A and serotonin secretes with the activation of platelet. And this causes vasospasm and agglutination and aggregation to platelet. This event again contributes increasing of the endothelium damage and this vicious circle goes on same. Anemia is microangiopatic hemolytic anemia. The erythrocytes whose endothelium is damaged and passing in little veins break and so fibrin deposits caused the little embolism. This embolism occurs in liver sinusoidal, perfusion of liver is out of order and ischemic necrosis improves. Thrombocytopenia depends on increasing demolishing platelet. The rate of maternal mortalite depends on different factors which are between 2% and 16.7%. Cerebral hemorrhage, disseminate intravascular hemolysis, acute respiratory distress syndrome, the deficiency of kidney, sepsis, hepatitis coma and ischemic encephalopathy are the most important causes of mortality. Classify of Mississippi which determines prognosis is classify is done according to the platelet counts. Type 1: Lower than 50,000/microL, Type 2: between 50,000 - 10,000/microL, Type 3: between 100,000 - 150,000 are accepted [26]. Treatment is form of supportive measures (if the fetus reaches enough size, pregnancy is terminated, replace of liquid, if there is infections, it is treated or hypertension treatment etc.). Especially in class 1 patient groups postpartum plasmapheresis prevents to morbidity and mortality was seen [27].
Myelodysplastic syndrome
Myelodysplastic syndrome (MDS) is characterized proliferation of myeloblastic leukemia cell and with the deficiency of bone marrow is a heterogeneous clonal group of hematology disease. There are cytopenia in peripheral blood and hyperplasia in bone marrow. The average age is 70. Although the pancytopenia has more seen in MDS patient, it can be apply with insulated thrombocytopenia. Patients according to the type of cytopenia can apply with repeating infection, bleeding, easy bruising, progressive weakness and lethargy or exercise dyspne. Thrombocytopenia has been seen approximately in half of the patients and it can be only a cytopenia among 5% percent. Qualitative and quantitative defects in platelets are reason precursor lesion like that petechia, purpura etc. and even it threats the life of bleeding [28, 29]. The most important bone marrow symptom in MDS patients is the myeloblast and its percent. If the rate of myeloblast is over 20% percent, it diagnoses as an acute myeloid leukemia. Searching bone marrow there has been often seen little micromegakaryocytes and most of the time lobulation abnormals is seen. Treatment of MDS the main strategy is to extend remaining alive. This strategy includes delaying the transformation to leukemia, decreasing of need for transfusion, reducing the infections and controlling, hematologic recovery and increasing quality of life. Only treatment method which knows for long term survival is allogenic bone marrow transplantation [30].
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria (PNH), is a rare, acquired, clonal, potentially life-threatening disease of the blood characterised by complement-induced intravascular hemolysis, cytopenia, red urine and thrombosis [31]. There is a somatic mutation in hemopoietic stem cell and affects the three cell sequence of hematopoietic system. With the help of GPI some of the proteins which connect to cell membrane is the deficiency mature cell is product of mutant hematopoietic stem cell [32]. Mostly the application of patients to the doctor is pancytopenia especially symptoms which are related to anemia patients. The cause of application in some patients is sudden the dark color of urine. Rarely first clinical symptom is thrombotic events which occur in venous system. In order to diagnose of the disease firstly the doctor should remember the PNH. In such clinical symptom of thrombosis, cytopenia, predisposition to thrombosis should be warning. Classical case of PNH is always expected findings. For that reason, coombs test negative or more correctly, there are no obvious signs of infections or schistocytes in non-spherocytic hemolytic anemia it should be suspected that PNH whether accompanied by hemoglobinuria or not. It is important to evaluate the clone of PNH during diagnosis in AA cases. It is the demonstration of the lack of CD55 and CD59 depended on GPl and fluorescein-labeled proaerolysin (FLAER) test in gold standard erythrocytes and leukoyctes for diagnosis in stream cytometry [30, 31]. It has been shown that corticosteroids have been reduced hemolysis in treatment. Eculizumab and complement inhibitor therapy that have been approved by FDA and shown to reduce the hemolysis and for the result of that the side effects of it in PNH cases are in the other treatment choice. Allogeneic stem cell transplantation is the only treatment that can provide curing in PNH in today [33].
Cyclic thrombocytopenia
It is regarded as a presentation of immune thrombocytopenia that are not mutad with the reason of it is not known. It improves as thrombocytopenia spontan that has been seen from time to time. It is mentioned menstrual cyclic thrombocytopenia that has been seen with the beginning of mens and occuring with the number of trombosit has become normal in the middle of cycle in some young women. The disease abates spontaneously after 3 - 7 episodes. It is thought that reducing the cyclic has a role in the construction of trombosit in the result of autoimmune platelet destruction in some patients and reducing trombopoetin cyclic in some patients. As a result of a cyclic increase in M-CSF, increasing thrombocyte phagocytosis can be also responsible for that. It can be seen both postmenopausal women and man. All treatment methods are ineffective. Birth control drugs can be helpful [34, 35].
Posttransfusion purpura
Posttransfusion Purpura (PTP) is very rare but severe complication of blood transfusion, characterised by the sudden onset of severe thrombocytopenia within 5 - 10 days of transfusion of blood products [36]. The blood transfusion starts an anamnestic response in raise HPA (human platelet specific alloantigen) antibodies in an already sensitised person. Thrombocytopenia in PTP is caused by antibody mediated demolition of both donor platelets as well as patient’s own platelets. The most commonly implicated platelet antibody responsible for PTP is anti-HPA-1a in an HPA-1a negative (HPA-1b/1b) patient receiving red cells or platelet concentrates from an HPA-1a positive donor [37]. Thrombocytopenia develops one week later after transufision. It becomes normal in 3 - 4 weeks. Plasmapheresis can be made if there are serious bleeding and intravenous immunoglobulin can be given [36, 38].
Chemotherapy-induced thrombocytopenia
With the increasing incidence of cancer, chemotherapy has become a common cause of thrombocytopenia. The history is usually readily available and patients often have cytopenia in other cell lines also. With most chemotherapy agents, rarely blood count occurs 7 to 10 days after chemotherapy and recovery over 2 to 3 weeks. Platelet transfusions are occasionally needed and dose adjustment of next chemotherapy doses may be necessary [4].
A suggested algorithm for evaluation of thrombocytopenia is presented in the Figure 1.
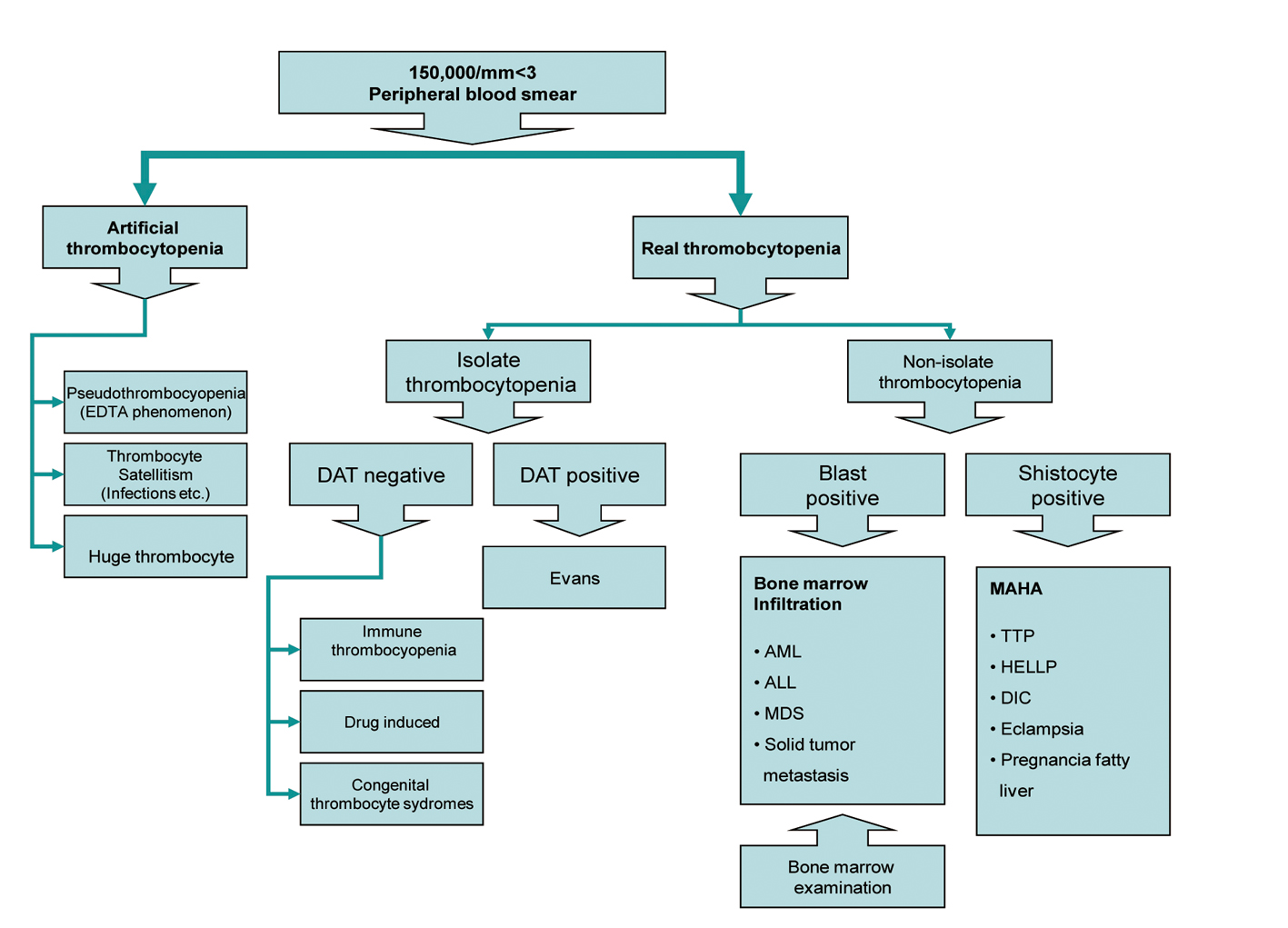 Click for large image | Figure 1. Algorithm for management of thrombocytopenia. |
| References | ▴Top |
- Levine SP. Thrombocytopenia: Pathophysiology and Classification.Ed.Greer JP, Foerster J, Rodgers GM, Paraskevas F, Glader B,Arber DA,Means RT. Wintrobe’s Clinical Hematology.12th ed. Lippincott Williams & Wilkins Co. Philadelphia. 2009; pp 1289-1334.
- Consolini DM. Thrombocytopenia in infants and children. Pediatr Rev. 2011;32(4):135-149; quiz 150-131.
pubmed - Goldstein KH, Abramson N. Efficient diagnosis of thrombocytopenia. Am Fam Physician. 1996;53(3):915-920.
pubmed - Sekhon SS, Roy V. Thrombocytopenia in adults: A practical approach to evaluation and management. South Med J. 2006;99(5):491-498; quiz 499-500, 533.
pubmed - Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Jr., Crowther MA. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190-4207.
pubmed doi - Doyle B, Porter DL. Thrombocytopenia. AACN Clin Issues. 1997;8(3):469-480.
pubmed doi - Haznedaroglu IC, Goker H, Turgut M, Buyukasik Y, Benekli M. Thrombopoietin as a drug: biologic expectations, clinical realities, and future directions. Clin Appl Thromb Hemost. 2002;8(3):193-212.
pubmed doi - Yildiz BO, Haznedaroglu IC, Coplu L. Albendazole-induced amegakaryocytic thrombocytopenic purpura. Ann Pharmacother. 1998;32(7-8):842.
pubmed doi - Schmidt KG, Rasmussen JW. Kinetics and distribution in vivo of 111In-labelled autologous platelets in idiopathic thrombocytopenic purpura. Scand J Haematol. 1985;34(1):47-56.
pubmed doi - Erkurt MA, Kaya E, Kuku I, Koroglu M, Aydogdu I. Management of Adult Immune Thrombocytopenia: Review Article. Journal of Inonu University Medical Faculty 2011,18(3): 203-212.
- Kaya E, Erkurt MA, Aydogdu I, Kuku I, Ozhan O, Oner RI, Ulutas O. Retrospective analysis of patients with idiopathic thrombocytopenic purpura from Eastern Anatolia. Med Princ Pract. 2007;16(2):100-106.
pubmed doi - Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, Chong BH, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115(2):168-186.
pubmed doi - George JN, Rizvi MA. Thrombocytopenia. Eds: Beutler E, Lichtman MA, Coller BS, Kipps TJ, Seligsohn A. Williams Hematology.6th ed. McGraw-Hill Co. New York. 2001; pp 1495-1539.
- Warkentin TE, Levine MN, Hirsh J, Horsewood P, Roberts RS, Gent M, Kelton JG. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med. 1995;332(20):1330-1335.
pubmed doi - Chong BH. Heparin-induced thrombocytopenia. Br J Haematol. 1995;89(3):431-439.
pubmed doi - Hunter JB, Lonsdale RJ, Wenham PW, Frostick SP. Heparin induced thrombosis: an important complication of heparin prophylaxis for thromboembolic disease in surgery. BMJ. 1993;307(6895):53-55.
pubmed doi - Warkentin TE. Heparin-induced thrombocytopenia: pathogenesis and management. Br J Haematol. 2003;121(4):535-555.
pubmed doi - Kajiwara E, Akagi K, Azuma K, Onoyama K, Fujishima M. Evidence for an immunological pathogenesis of thrombocytopenia in chronic liver disease. Am J Gastroenterol. 1995;90(6):962-966.
pubmed - Hoofnagle JH. Thrombocytopenia during interferon alfa therapy. JAMA. 1991;266(6):849.
pubmed doi - Ingeberg S, Stoffersen E. Platelet dysfunction in patients with vitamin B12 deficiency. Acta Haematol. 1979;61(2):75-79.
pubmed doi - Young NS, Issaragrasil S, Chieh CW, Takaku F. Aplastic anaemia in the Orient. Br J Haematol. 1986;62(1):1-6.
pubmed doi - Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
pubmed - Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585-1594.
pubmed doi - Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325(6):398-403.
pubmed doi - Padden MO. HELLP syndrome: recognition and perinatal management. Am Fam Physician. 1999;60(3):829-836, 839.
pubmed - Martin JN, Jr., Blake PG, Lowry SL, Perry KG, Jr., Files JC, Morrison JC. Pregnancy complicated by preeclampsia-eclampsia with the syndrome of hemolysis, elevated liver enzymes, and low platelet count: how rapid is postpartum recovery? Obstet Gynecol. 1990;76(5 Pt 1):737-741.
pubmed doi - Julius CJ, Dunn ZL, Blazina JF. HELLP syndrome: laboratory parameters and clinical course in four patients treated with plasma exchange. J Clin Apher. 1994;9(4):228-235.
pubmed doi - Aul C, Gattermann N, Schneider W. Age-related incidence and other epidemiological aspects of myelodysplastic syndromes. Br J Haematol. 1992;82(2):358-367.
pubmed doi - Heaney ML, Golde DW. Myelodysplasia. N Engl J Med. 1999;340(21):1649-1660.
pubmed doi - Hellstrom-Lindberg E. Efficacy of erythropoietin in the myelodysplastic syndromes: a meta-analysis of 205 patients from 17 studies. Br J Haematol. 1995;89(1):67-71.
pubmed - Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253-1258.
pubmed doi - Nakakuma H, Nagakura S, Iwamoto N, Kawaguchi T, Hidaka M, Horikawa K, Kagimoto T, et al. Paroxysmal nocturnal hemoglobinuria clone in bone marrow of patients with pancytopenia. Blood. 1995;85(5):1371-1376.
pubmed - Parker CJ. Management of paroxysmal nocturnal hemoglobinuria in the era of complement inhibitory therapy. Hematology Am Soc Hematol Educ Program. 2011;2011:21-29.
pubmed - Balduini CL, Stella CC, Rosti V, Bertolino G, Noris P, Ascari E. Acquired cyclic thrombocytopenia-thrombocytosis with periodic defect of platelet function. Br J Haematol. 1993;85(4):718-722.
pubmed doi - Go RS. Idiopathic cyclic thrombocytopenia. Blood Rev. 2005;19(1):53-59.
pubmed doi - Warkentin TE, Smith JW. The alloimmune thrombocytopenic syndromes. Transfus Med Rev. 1997;11(4):296-307.
pubmed doi - Lucas GF, Pittman SJ, Davies S, Solanki T, Bruggemann K. Post-transfusion purpura (PTP) associated with anti-HPA-1a, anti-HPA-2b and anti-HPA-3a antibodies. Transfus Med. 1997;7(4):295-299.
pubmed doi - Santoso S, Kiefel V. Human platelet alloantigens. Wien Klin Wochenschr. 2001;113(20-21):806-813.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.