

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Review
Volume 11, Number 4, August 2022, pages 123-130

Treatment of Light Chain Deposition Disease: A Systematic Review
Adeel Masooda, f ![]() , Hamid Ehsanb, Qamar Iqbalc, Ahmed Salmand, Hamza Hashmie
, Hamid Ehsanb, Qamar Iqbalc, Ahmed Salmand, Hamza Hashmie
aHematology/Oncology, MD Anderson Cancer Center, Houston, TX 77030, USA
bHematology/Oncology, Levine Cancer Institute/Atrium Health, Charlotte, NC, USA
cInternal Medicine, Tidal Health Peninsula Regional, Salisbury, MD 21801, USA
dHematology/Oncology, Rochester Regional Health/Lipson Cancer Center, USA
eHematology/Oncology, Medical University of South Carolina, Charleston, SC 29425, USA
fCorresponding Author: Adeel Masood, Hematology/Oncology, MD Anderson Cancer Center, Houston, TX 77030, USA
Manuscript submitted July 23, 2022, accepted August 13, 2022, published online August 30, 2022
Short title: Review of Treatment of LCDD
doi: https://doi.org/10.14740/jh1038
| Abstract | ▴Top |
Light chain deposition disease (LCDD) is a rare hematologic disorder that can affect any organ but predominantly involves the kidneys. Existing literature is limited to case reports and small single-center retrospective series, explaining the lack of any treatment algorithms and management guidelines for patients with this disorder. In this systematic review of literature, we explored the role of standard and high-dose chemotherapy-autologous stem cell transplant for LCDD. A total of 11 studies were identified to evaluate the hematologic and renal responses to various treatment regimens. Autologous stem cell transplant and bortezomib-based regimens appear to have reasonable safety and efficacy for this rare hematologic disorder, albeit some statistical and analytical limitations. Large multicenter retrospective and prospective studies are needed to better elucidate the role of various chemotherapy regimens as well as autologous stem cell transplant for patients with LCDD.
Keywords: Light chain deposition disease; Bortezomib; Autologous stem cell transplant; Lenalidomide; Thalidomide
| Introduction | ▴Top |
Light chain deposition disease (LCDD) is a clonal plasma cell disorder characterized by the deposition of nonamyloid monoclonal light chains in multiple organs. It is a relatively rare condition with no known or reported incidence in literature. The median age at diagnosis is around 58 years, with a slightly higher preponderance for men [1]. More recently, International Kidney and Monoclonal Gammopathy Research Group Consensus characterized LCDD as part of monoclonal gammopathy of renal significance (MGRS), a broader spectrum of B-cell proliferative disorders known to create monoclonal immunoglobulins toxic to kidneys. MGRS is divided into organized (fibrillar, microtubular, crystalline, or inclusionary) and non-organized, with LCDD falling into the latter category [2]. In contrast to patients with multiple myeloma (MM), underlying pathophysiology in LCDD involves end-organ damage associated with abnormal deposition of light chains as produced by the plasma cells, regardless of the plasma cell burden or magnitude of light chains production. Because of this, the disease course in LCDD is often aggressive and is associated with an overall poor prognosis [3]. The immunoglobulin light chains in LCDD are predominantly kappa light chains with granular tissue deposits. Unlike AL amyloidosis, these light chains do not form amyloid fibrils or stain Congo red [4]. Immunofluorescence staining is an important step in the initial diagnosis of LCDD, which usually shows monotypic deposition of light chains [5]. LCDD is also categorized as a monoclonal immunoglobulin deposition disease (MIDD) in the World Health Organization (WHO) classification of plasma cell disorders. Other plasma cell disorders included in MIDD include heavy chain deposition disease (HCDD) and light heavy chain deposition disease (LHCDD), but LCDD is the most common entity amongst all [6, 7]. Factors associated with a worse prognosis include older age, advanced renal disease at the initial diagnosis, other associated plasma cell disorders, and extrarenal LCDD [8]. Due to the rarity of the disease and lack of randomized clinical trials, there are currently no Food and Drug Administration (FDA) approved therapies or universally accepted standard of care treatment options available for LCDD. The main goal of treatment is to slow the production and tissue deposition of light chains to prevent further damage to the organ function. The treatment scheme is similar to MM, primarily including proteasome inhibitors (bortezomib), immunomodulatory agents (lenalidomide, thalidomide), and autologous stem cell transplant (ASCT). This review aims to provide a systematic summary of available literature regarding the various treatment options, including safety and efficacy of standard and high-dose chemotherapy for LCDD.
| Methods | ▴Top |
A comprehensive literature search was performed on PubMed, Cochrane, EMBASE, and Clinicaltrials.gov. All reported cases of LCDD without any evidence of symptomatic MM by CRAB criteria (calcium elevation, renal dysfunction, anemia, and bone disease) were included in the qualitative analysis. Due to the rarity of the disease, we could not find any clinical trials evaluating LCDD. Therefore, retrospective single-center reports/case series were included in the qualitative analysis. Cases of LCDD in transplanted kidneys were excluded since it was not possible to determine whether the disease process started before or after the kidney transplant. However, patients that received a kidney transplant after diagnosis and treatment for LCDD were included in the analysis. Any patient who did not receive treatment was also excluded (Fig. 1).
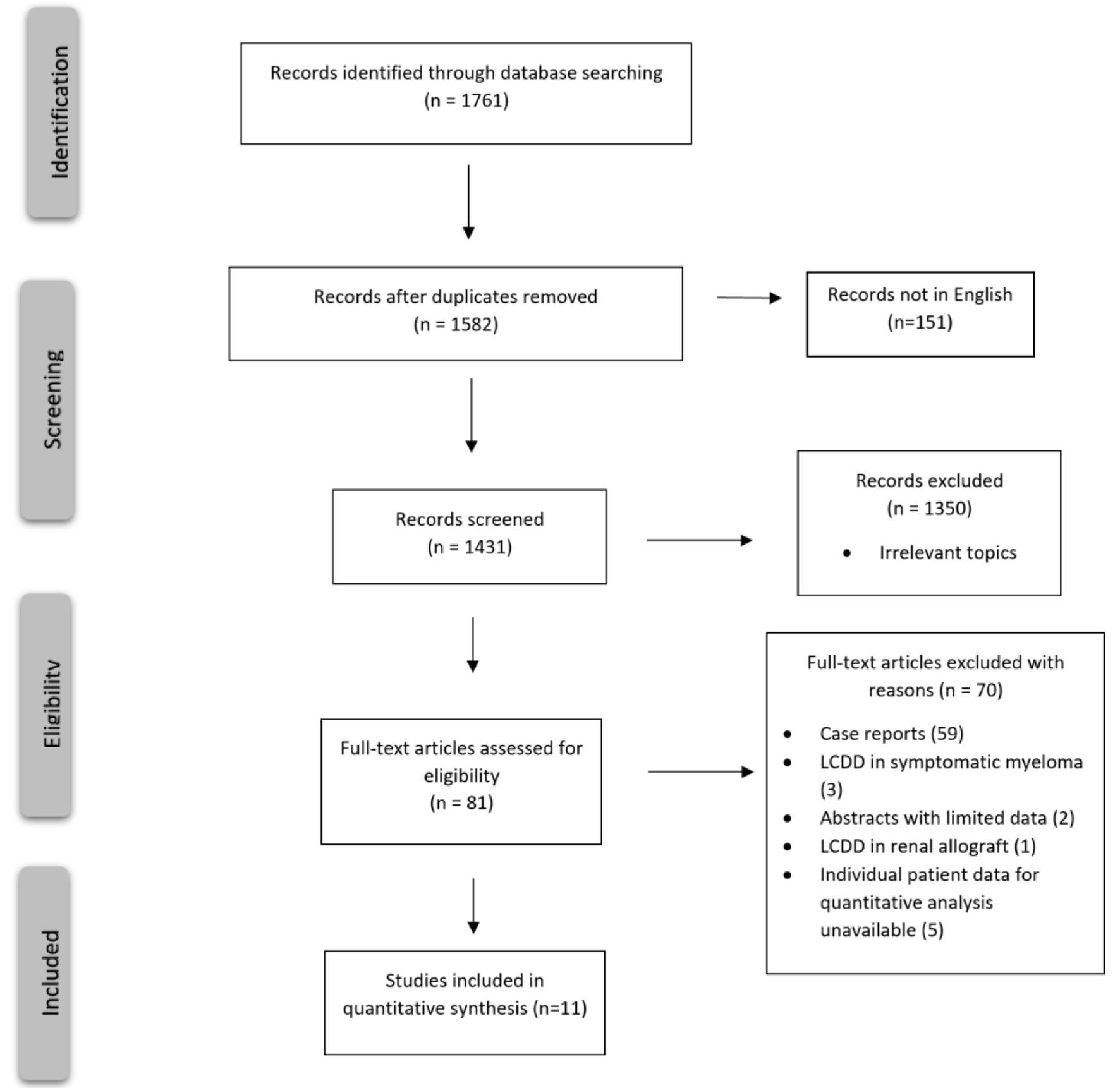 Click for large image | Figure 1. PRISMA flow chart for study selection criteria. |
Amyloidosis response criteria were used to determine hematologic or renal responses if not reported in the study, depending on the availability of relevant laboratory data [9]. Serum and urine monoclonal proteins by immunofixation and kappa/lambda (K/L) free light chains and ratios were used to determine the hematologic response. Similarly, the renal responses were determined based on the availability of the results of 24-h urine samples before and after treatment, if not reported in the study.
JMP Pro 16 [10] was used for the descriptive statistics and the pooled analysis. Due to the rarity of the disease and skewed data (outliers in data), medians with range were used to report interval/ratio variables. Frequencies and percentages were used for the categorical variables. Some studies reported serum creatinine in µmol/L that was converted to mg/dL using an online calculator to ascertain the uniformity of data.
| Results | ▴Top |
This systematic review evaluated various treatment regimens for LCDD, such as ASCT, bortezomib-based regimens, and thalidomide/lenalidomide-based regimens. Out of the initial 81 studies identified, 70 were excluded because they were either case reports or did not report individual patients’ characteristics and did not isolate idiopathic LCDD from symptomatic MM and other hematologic malignancies (Fig. 1). Subsequently, 11 studies (n = 64) were included in this systematic review (Table 1 [11-22]).
 Click to view | Table 1. Comparison of Hematologic and Organ Responses With Various Regimens for Light Chain Deposition Disease |
The pooled analysis revealed a complete response (CR) rate of 61.5% with the bortezomib-dexamethasone (BorD) induction chemotherapy followed by ASCT (n = 8/13). Bortezomib-based regimens without subsequent ASCT were associated with a CR rate of 55.6% (n = 10/18). Similarly, thalidomide (CR 27%; n = 3/11) and lenalidomide (75% very good partial response (VGPR); n = 3/4) based regimens appeared to have comparable efficacy to bortezomib-based regimens. As far as the renal responses are concerned, 84% of the patients (n = 21/25) undergoing ASCT (with or without bortezomib or other prior induction therapies) had more than 50% decrease in 24-h proteinuria (partial renal response based on amyloidosis response criteria) [9]. Only five patients undergoing bortezomib-based regimens had organ response data available, and 60% achieved a partial response (PR) (n = 3/5). While response data were not reported for any of the 11 patients receiving thalidomide-based regimens, none of the patients in the response evaluable lenalidomide group achieved an organ response. Renal (n = 33) and hematologic responses (n = 61) are summarized as bar charts below (Figs. 2 and 3). It is of note that hematologic responses were not reported or evaluable in 3/64 patients and renal responses were not evaluable in 31/64 patients.
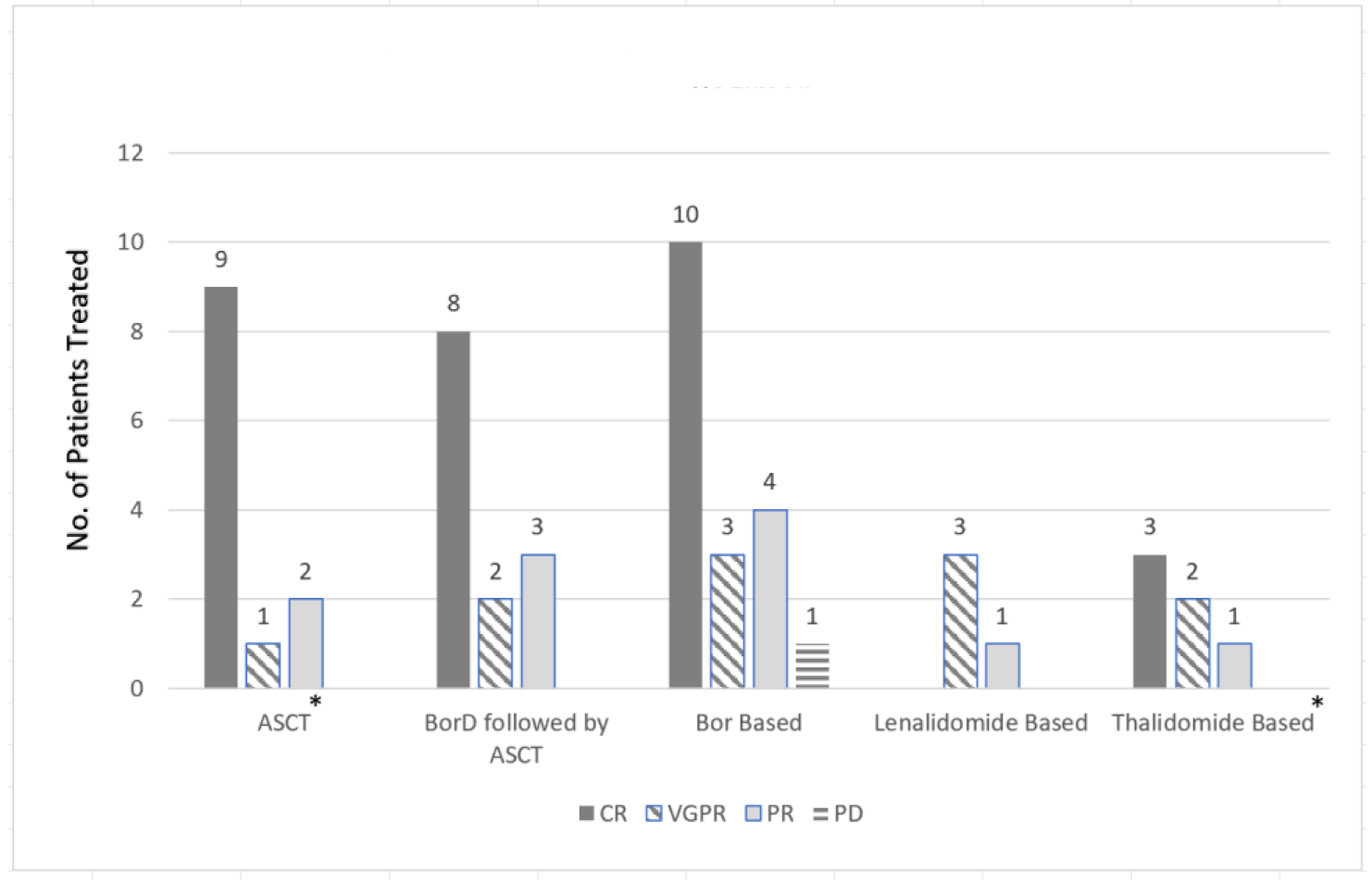 Click for large image | Figure 2. Hematological response comparison based on treatment regimen. *No response patients not included in the graph: three patients with ASCT and five patients with thalidomide-based therapies. ASCT: autologous stem cell transplant; BorD: bortezomib and dexamethasone; Bor: bortezomib; no.: number; CR: complete response; VGPR: very good partial response; PR: partial response; PD: progression of disease. |
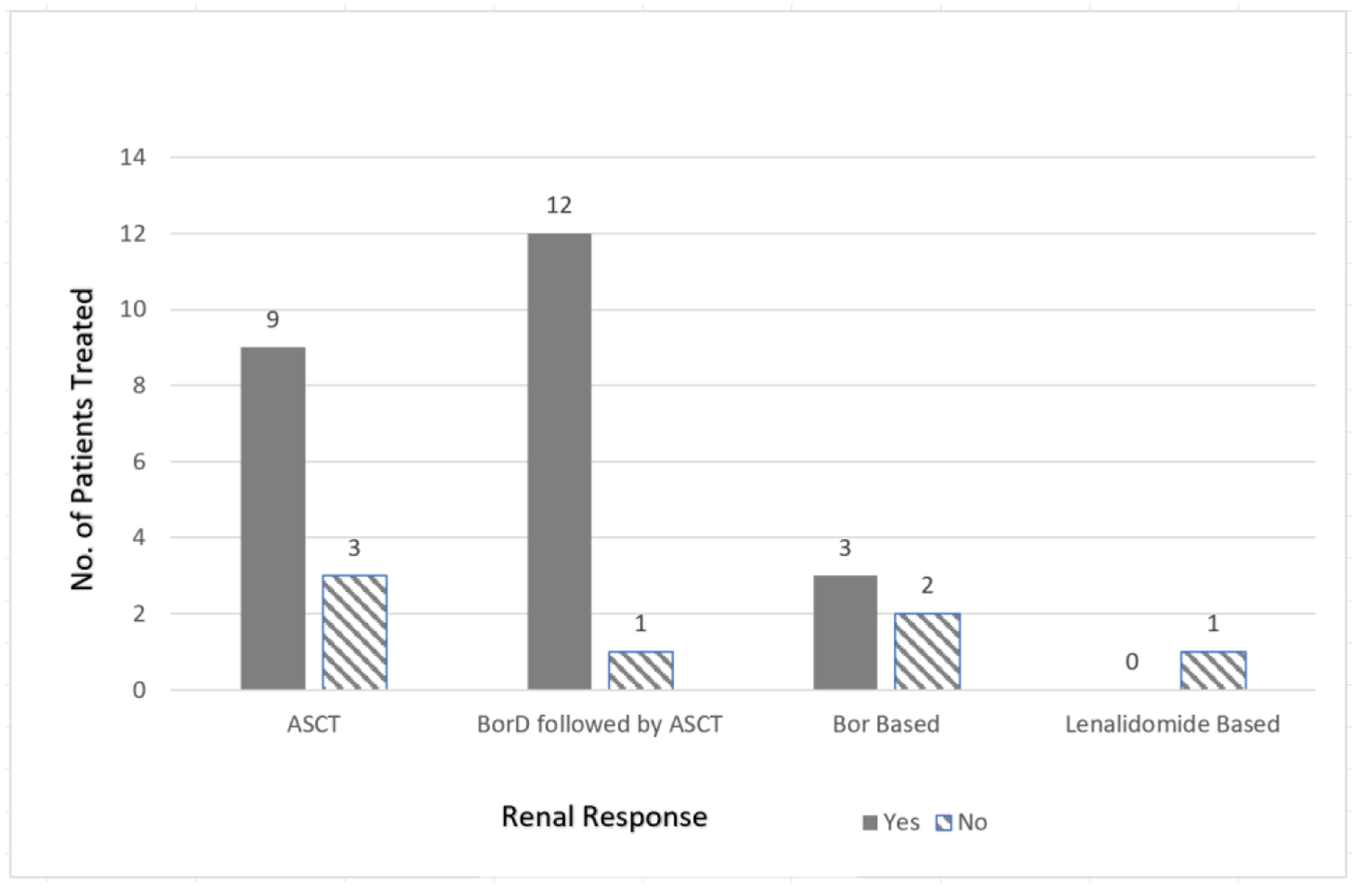 Click for large image | Figure 3. Renal response comparison based on treatment regimen. No.: number; ASCT: autologous stem cell transplant; BorD: bortezomib and dexamethasone; Bor: bortezomib-based regimens. |
Pooled analysis based on K/L ratio and renal parameters before and after treatment was conducted for all those with evaluable data. The median K/L ratio before treatment was 32 (range: 0.001 - 252) for 31/64 evaluable patients. Similarly, after treatment, the median K/L ratio was 1.005 (range: 0.24 - 36.9) for 31/64 evaluable patients. The median serum creatinine before treatment was 2.25 mg/dL (range: 0.89 - 5.4) for 30/64 evaluable patients and was 1.7 mg/dL (range: 0.7 - 6.3) after treatment for the 21/64 evaluable patients. The median 24-h proteinuria before treatment was 4.25 g/day (range: 0.51 - 15) for 32/64 evaluable patients, and 1.17 g/day (range: 0.173 - 5.9) for the 24/64 evaluable patients afterwards.
Since a majority of patients (72% (n = 46/64)) underwent bortezomib-based regimens or ASCT (with/without the mention of induction therapy), pooled analysis of renal parameters and K/L ratio was done for these two cohorts separately. Eight out of 11 studies included in this analysis reported utilization of ASCT (n = 28), and four of the 11 studies reported bortezomib-based regimens (n = 18). ASCT appeared to yield better outcomes for renal responses compared to bortezomib-based regimens without ASCT, as the median 24-h urine protein in the ASCT group after treatment was 1.14 g/day (range: 0.173 - 5.9) compared to median 24-h urine protein of 2.3 g/day (range: 0.61 - 4.8) for bortezomib-based regimens without ASCT. On the contrary, the K/L ratio was not significantly different after treatment for both ASCT (median 1.005; range: 0.24 - 6.5) and bortezomib-only groups (median 0.93; range: 0.37 - 36.9).
Among individual studies, few are noteworthy such as Sayed et al, 2015 who reported the largest single-center retrospective analysis of idiopathic LCDD from the United Kingdom National Amyloidosis Center. The hematologic and renal parameters were not reported for the individual cases, but patients were stratified into groups based on treatment regimen and hematologic response. Out of the 25 evaluable patients, four underwent ASCT and achieved hematologic CR. Similarly, 89% of the patients in the bortezomib group (n = 8/9) and 27% of the patients in the thalidomide group (n = 3/11) achieved CR. Only one patient received lenalidomide and had a PR. Renal responses were not reported for any of the patients [17].
Lorenz et al, 2008 reported a single-center experience with long-term outcomes of three patients following ASCT. One patient achieved a renal response post-ASCT. Although hematologic responses were not reported, 2/3 of patients were alive at the time of the last follow-up (median follow-up 46.2 months; range: 33 - 43) [11].
Jimenez-Zepeda et al, 2012 reported another single-center experience with six patients with LCDD that underwent ASCT. Half of the patients (n = 3) received BorD while the other half received dexamethasone alone pre-ASCT. The study reported hematologic and renal responses (based on serum creatinine and 24-h proteinuria) post-induction and 6 months post-ASCT. All of the patients achieved a renal response post-transplant. Two-thirds of the patients achieved CR, 17% had a PR, and 17% had no response [13].
Kastritis et al, 2019 reported four patients from a single center who received BorD for induction chemotherapy prior to ASCT. Two out of four patients achieved hematologic CR. Three (75%) patients subsequently underwent ASCT. All patients achieved a renal response, and all patients were alive at the time of the last follow-up (range: 10 - 18 months). Based on this case series, bortezomib-based regimens seemed to be a reasonable option for patients prior to ASCT [14].
Kastritis et al, 2021 reported six LCDD patients who received a short 4-week consolidation course of daratumumab after eight cycles of bortezomib, cyclophosphamide, and dexamethasone (VCd). All patients had renal involvement at the time of diagnosis and failed to achieve a CR with VCd. Three patients (50%) were already in hematologic VGPR, and the rest (50%) were in PR before daratumumab initiation. After four weekly daratumumab administrations, one patient improved to hematologic CR, while three remained in VGPR, and the rest of the two stayed in PR. The free light chain (FLC) ratio normalized in 50% of patients after the consolidation as opposed to none prior. Only noticeable adverse event was mild injection-related reaction, and no hematological adverse event was reported [22].
Due to the rarity of idiopathic LCDD, most large studies combine similar diagnoses to make a meaningful interpretation of the efficacy and safety of various treatment regimens. One nationwide French study by Joly et al, 2019 reported 255 patients with MIDD (60% idiopathic LCDD, 23% LCDD with cast nephropathy, and 17% HCDD/LHCDD). Of note, 34% of patients had symptomatic myeloma, 64% had MGRS, and 35% had hepatic or cardiac involvement. Overall, 66% of patients (n = 169/255) were treated with chemotherapy (58% bortezomib-based regimens, 17% alkylating agents and 10% immunomodulatory agents), while 15% (n = 38/255) were treated with high-dose chemotherapy-ASCT. On univariate analysis, both ASCT and bortezomib-based regimens had a statistically significant hematologic response. Interestingly, the median overall survival (OS) was found to be 140 months with idiopathic LCDD compared to 28 months for LCDD with cast nephropathy [23]. Another recent multicenter retrospective observational study compared outcomes in biopsy-proven amyloidosis (n = 180) and non-amyloidosis-associated MGRS (n = 100; various etiologies). MIDD constituted 53 patients in this study. Most patients received proteasome inhibitors followed by conventional chemotherapy as first-line treatment. Overall, non-amyloidosis-associated MGRS was found to have an overall response rate (ORR) of 72% compared to 56% in amyloidosis-associated disease. VGPR or better hematologic response was associated with a better probability of renal response in the non-amyloidosis MGRS group (renal response of 77% vs. 47% with ≥ VGPR vs. PR/stable disease hematologic response, respectively) [24]. Both these studies were not included in our quantitative analysis as individual data and outcomes for study participants or subgroups were not reported separately.
| Discussion | ▴Top |
Plasma cell disorders, including myeloma cast nephropathy, cryoglobulinemia, AL amyloidosis, and LCDD, can be associated with renal impairment with clearly defined pathophysiological mechanisms. In LCDD, granular light chains produced by clonal plasma cells in the bone marrow are deposited in glomeruli causing characteristic nodular glomerulosclerosis [25]. In contrast to other plasma cell disorders, including AL amyloidosis, LCDD typically only affects kidneys and, in some rare instances, the heart and liver. Light chains are typically kappa-restricted, and there is usually a monoclonal plasma cell population in the bone marrow, albeit a small amount. The disorder is more commonly seen in males that are relatively younger than patients with MM or amyloidosis [1]. Treatment strategies comprise systemic chemotherapy with or without ASCT to eliminate plasma cell burden in the bone marrow that produces light chains. This leads to long-term hematologic remission and improvement in organ function, reflected by improvement in estimated glomerular filtration rate, serum creatinine, and proteinuria. As highlighted in our review, bortezomib-based therapy appears to be favorable as no dose modification is required for renal insufficiency, and early hematologic responses can be seen that translate to the recovery of renal function. Similarly, high-dose chemotherapy-ASCT can achieve durable hematologic remission and long-term disease control [11, 20]. One recent clinical trial of daratumumab in eight LCDD patients with a concomitant diagnosis of MM showed reasonable hematologic and renal responses (50% achieved VGPR and 25% achieved renal response), warranting further investigations into monoclonal antibodies [26]. Although it appears that the patients undergoing ASCT seem to achieve deeper and more durable hematologic remissions and organ responses, no statistically significant superiority can be claimed over non-transplant or standard chemotherapy-based approaches. Patient selection is a source of potential bias here as patients with adequate performance status and organ function, especially renal reserve, are likely to undergo ASCT. It is important to highlight that renal/organ recovery is not always guaranteed, and according to some experts, if a patient with LCDD is not deemed a candidate for renal transplant, the role of systemic chemotherapy remains unclear unless there is evidence of hepatic or cardiac infiltration [27]. For patients that are, in fact, candidates for renal allograft, control of light chain production is necessary to avoid ongoing damage to the transplanted kidney [28]. Due to the rarity of the disorder, a small number of patients, lack of registry data, and details on hematologic/renal responses, the role, timing, and pre-/post-ASCT strategies remain unclear.
There are several limitations associated with this systematic review. In the absence of a conclusive diagnostic renal biopsy in some cases, it is difficult to distinguish whether renal impairment was due to LCDD versus cast nephropathy caused by MM or renal damage due to beta-pleated light chains in AL amyloidosis. All of the studies included in the analysis are retrospective in nature and had a small number of patients. Moreover, hematologic and organ responses were either not reported or differed in response evaluation criteria used in the various studies in our analysis. This is a source of potential bias making the cross-study comparison of various chemotherapeutic options challenging. Follow-up remains short, making it difficult to draw meaningful conclusions about the long-term efficacy of standard or high-dose chemotherapy for patients with LCDD. Larger retrospective analyses using registry data like Center for International Blood and Marrow Transplant (CIBMTR) and or randomized multicenter retrospective/prospective studies with clearly defined hematologic and organ response evaluation criteria are needed to determine the safety and efficacy of the various treatment regimens and modalities available for this disorder.
| Conclusion | ▴Top |
LCDD is a rare hematologic disorder, with renal impairment being the primary morbidity associated with this disorder. In the eligible patient population, bortezomib-based chemotherapy followed by ASCT appears to be an effective treatment option with durable hematologic remission and organ responses. While early recognition and timely therapy may help with the recovery of organ function, large multicenter retrospective and prospective studies are needed to elucidate the role, sequence, and timing of various chemotherapy regimens as well as ASCT for patients with LCDD.
Acknowledgments
None to declare.
Financial Disclosure
The authors received no funding for this project.
Conflict of Interest
Authors do not have any conflict of interest to report.
Author Contributions
AM, HE, and HH contributed to the study design. AM and QI did the literature search. AM, QI, HE, AS, and HH wrote the manuscript. HH did the expert review of the final manuscript. The final version of the manuscript has been approved by all authors who have agreed to be accountable for all aspects of the work.
Data Availability
The authors declare that data supporting the findings of this study are available from the corresponding author.
| References | ▴Top |
- Pozzi C, D'Amico M, Fogazzi GB, Curioni S, Ferrario F, Pasquali S, Quattrocchio G, et al. Light chain deposition disease with renal involvement: clinical characteristics and prognostic factors. Am J Kidney Dis. 2003;42(6):1154-1163.
doi pubmed - Leung N, Bridoux F, Batuman V, Chaidos A, Cockwell P, D'Agati VD, Dispenzieri A, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019;15(1):45-59.
doi pubmed - Dhodapkar MV, Merlini G, Solomon A. Biology and therapy of immunoglobulin deposition diseases. Hematol Oncol Clin North Am. 1997;11(1):89-110.
doi - Buxbaum JN, Chuba JV, Hellman GC, Solomon A, Gallo GR. Monoclonal immunoglobulin deposition disease: light chain and light and heavy chain deposition diseases and their relation to light chain amyloidosis. Clinical features, immunopathology, and molecular analysis. Ann Intern Med. 1990;112(6):455-464.
doi pubmed - Ronco P, Bridoux F, Aucouturier P. Monoclonal gammopathies: Multiple myeloma, amyloidosis, and related disorders. Diseases of the Kidney 2: Little Brown and Company Boston. 1996. p. 2129-2174.
- Kanzaki G, Okabayashi Y, Nagahama K, Ohashi R, Tsuboi N, Yokoo T, Shimizu A. Monoclonal immunoglobulin deposition disease and related diseases. J Nippon Med Sch. 2019;86(1):2-9.
doi pubmed - Kourelis TV, Nasr SH, Dispenzieri A, Kumar SK, Gertz MA, Fervenza FC, Buadi FK, et al. Outcomes of patients with renal monoclonal immunoglobulin deposition disease. Am J Hematol. 2016;91(11):1123-1128.
doi pubmed - Buxbaum J, Gallo G. Nonamyloidotic monoclonal immunoglobulin deposition disease. Light-chain, heavy-chain, and light- and heavy-chain deposition diseases. Hematol Oncol Clin North Am. 1999;13(6):1235-1248.
doi - International Myeloma Foundation. N.D. International myeloma working group uniform response criteria for multiple myeloma. Retrieved on Nov. 24th, 2021. Available from: https://www.myeloma.org/resource-library/international-myeloma-working-group-imwg-uniform-response-criteria-multiple.
- p. JMP® Version 16. SAS Institute Inc., Cary, NC. 1989-2021.
- Lorenz EC, Gertz MA, Fervenza FC, Dispenzieri A, Lacy MQ, Hayman SR, Gastineau DA, et al. Long-term outcome of autologous stem cell transplantation in light chain deposition disease. Nephrol Dial Transplant. 2008;23(6):2052-2057.
doi pubmed - Kimura S, Ohkawara H, Ogawa K, Tanaka M, Sano T, Harada-Shirado K, Takahashi H, et al. Lenalidomide as a Beneficial Treatment Option for Renal Impairment Caused by Light Chain Deposition Disease. Intern Med. 2018;57(24):3651-3657.
doi pubmed - Jimenez-Zepeda VH, Trudel S, Winter A, Reece DE, Chen C, Kukreti V. Autologous stem cell transplant for light chain deposition disease: incorporating bortezomib to the induction therapy. Am J Hematol. 2012;87(8):822-823.
doi pubmed - Kastritis E, Migkou M, Gavriatopoulou M, Zirogiannis P, Hadjikonstantinou V, Dimopoulos MA. Treatment of light chain deposition disease with bortezomib and dexamethasone. Haematologica. 2009;94(2):300-302.
doi pubmed - Lessi F, Castelli M, Trentin L, Altinier S, Piazza F, Fausto A. Bortezomib-dexamethasone as induction therapy for light chain deposition disease (LCDD): A single center experience. Blood. 2012;120(21):5027.
doi - Minarik J, Scudla V, Tichy T, Pika T, Bacovsky J, Lochman P, Zadrazil J. Induction treatment of light chain deposition disease with bortezomib: rapid hematological response with persistence of renal involvement. Leuk Lymphoma. 2012;53(2):330-331.
doi pubmed - Sayed RH, Wechalekar AD, Gilbertson JA, Bass P, Mahmood S, Sachchithanantham S, Fontana M, et al. Natural history and outcome of light chain deposition disease. Blood. 2015;126(26):2805-2810.
doi pubmed - Telio D, Shepherd J, Forrest D, Zypchen L, Barnett M, Nevill T, Song KW. High-dose melphalan followed by auto-SCT has favorable safety and efficacy in selected patients with light chain deposition disease and light and heavy chain deposition disease. Bone Marrow Transplant. 2012;47(3):453-455.
doi pubmed - Tovar N, Cibeira MT, Rosinol L, Sole M, de Larrea CF, Escoda L, Rovira M, et al. Bortezomib/dexamethasone followed by autologous stem cell transplantation as front line treatment for light-chain deposition disease. Eur J Haematol. 2012;89(4):340-344.
doi pubmed - Weichman K, Dember LM, Prokaeva T, Wright DG, Quillen K, Rosenzweig M, Skinner M, et al. Clinical and molecular characteristics of patients with non-amyloid light chain deposition disorders, and outcome following treatment with high-dose melphalan and autologous stem cell transplantation. Bone Marrow Transplant. 2006;38(5):339-343.
doi pubmed - Hassan Zafar S, Sinha S, Bhargava M. Spectrum of monoclonal light chain gammopathies: A 5 year institutional experience. Indian Journal of Hematology and Blood Transfusion. 2011;27(4):221-222.
- Kastritis E, Rousakis P, Kostopoulos IV, Gavriatopoulou M, Theodorakakou F, Fotiou D, Dialoupi I, et al. Consolidation with a short course of daratumumab in patients with AL amyloidosis or light chain deposition disease. Amyloid. 2021;28(4):259-266.
doi pubmed - Joly F, Cohen C, Javaugue V, Bender S, Belmouaz M, Arnulf B, Knebelmann B, et al. Randall-type monoclonal immunoglobulin deposition disease: novel insights from a nationwide cohort study. Blood. 2019;133(6):576-587.
doi pubmed - Gozzetti A, Guarnieri A, Zamagni E, Zakharova E, Coriu D, Bittrich M, Pika T, et al. Monoclonal gammopathy of renal significance (MGRS): Real-world data on outcomes and prognostic factors. Am J Hematol. 2022;97(7):877-884.
doi pubmed - Preud'homme JL, Aucouturier P, Touchard G, Striker L, Khamlichi AA, Rocca A, Denoroy L, et al. Monoclonal immunoglobulin deposition disease (Randall type). Relationship with structural abnormalities of immunoglobulin chains. Kidney Int. 1994;46(4):965-972.
doi pubmed - Milani P, Basset M, Curci P, Foli A, Rizzi R, Nuvolone M, Guido R, et al. Daratumumab in light chain deposition disease: rapid and profound hematologic response preserves kidney function. Blood Adv. 2020;4(7):1321-1324.
doi pubmed - Gertz MA. Managing light chain deposition disease. Leuk Lymphoma. 2012;53(2):183-184.
doi pubmed - Leung N, Lager DJ, Gertz MA, Wilson K, Kanakiriya S, Fervenza FC. Long-term outcome of renal transplantation in light-chain deposition disease. Am J Kidney Dis. 2004;43(1):147-153.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.