

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 11, Number 2, April 2022, pages 66-70

An Unusual Case of Delayed Hemolytic Transfusion Reaction With Hyperhemolysis Syndrome Due to Anti-Jkb and Anti-Fya Alloantibodies
Kenza El Alaouia, b ![]() , Fleur Samantha Benghiata, Martin Colarda
, Fleur Samantha Benghiata, Martin Colarda
aDepartment of Hematology, Erasme Hospital, Universite Libre de Bruxelles, Brussels, Belgium
bCorresponding Author: Kenza El Alaoui, Department of Hematology, Erasme Hospital, Universite Libre de Bruxelles, Brussels, Belgium
Manuscript submitted January 14, 2022, accepted February 22, 2022, published online April 12, 2022
Short title: DHTR Without Hematologic Disorder
doi: https://doi.org/10.14740/jh968
| Abstract | ▴Top |
Delayed hemolytic transfusion reaction (DHTR) is a complication appearing a few days to weeks due to alloimmunization following packed red blood cells (RBCs) transfusion, a pregnancy, or transplantation. Hyperhemolysis syndrome (HS) is a severe form of DHTR defined by a drop of hemoglobin to a level lower than before the transfusion, reflecting a destruction of the patient’s own RBCs not presenting the targeted antigen as well as the transfused RBCs. Usually seen in sickle cell disease (SCD) patients, HS remains very rare in patients without a hematologic disorder. We report the case of an 82-year-old Caucasian woman who presented with a DHTR with HS after being transfused packed RBC twice in the context of rectal bleeding. The patient was not known for any hemoglobinopathy and did not have a history of massive transfusions nor multiple pregnancies putting her at risk of alloimmunization. Our patient developed anti-C, anti-Fya and anti-Jkb antibodies, known to be harmful antibodies. First line of treatment after avoidance of further transfusions is intravenous immunoglobulins for 3 to 5 days and high-dose corticosteroids. Exceptional in the non-SCD population, this complication should be recalled by clinicians as it can be fatal if not treated appropriately. We performed a review of the literature using the words “delayed hemolytic transfusion reaction” and “hyperhemolysis syndrome” for similar cases. Finally, we describe how to diagnose, manage, and prevent this potentially fatal complication, which is still underrecognized even within the SCD population.
Keywords: Delayed hemolytic transfusion reaction; Hyperhemolysis syndrome; Diagnose; Management
| Introduction | ▴Top |
Delayed hemolytic transfusion reaction (DHTR) is due to alloimmunization following packed red blood cells (RBCs) transfusion, a pregnancy, or transplantation. Due to antibody evanescence with time, further transfusions can induce, in an anamnestic response setting, a hemolytic reaction which appears a few days to weeks (average 9 days) after the transfusion. There is a significant drop of hemoglobin post transfusion, along with an important lactate dehydrogenase (LDH) rise and possible hemoglobinuria. Delayed serologic transfusion reaction, however, refers to the sole identification of an antibody without evidence of hemolysis [1].
DHTR is more common in sickle cell disease (SCD) patients [2, 3], as they are more prone to alloimmunization than other frequently transfused patients. Indeed, usual RBC cross-matching include the main antigens (ABO, RhD), and SCD patients often receive transfusions from blood donors of a different ethnicity, exposing them to different blood group antigens [4].
Hyperhemolysis syndrome (HS) is defined by a drop of hemoglobin to a level lower than before the transfusion, reflecting a destruction of the transfused RBCs with the autologous RBCs [5]. This more severe form of DHTR can lead to life-threatening anemia and end organ damage, requiring immediate management.
| Case Report | ▴Top |
Investigations
We report the case of an 82-year-old Caucasian woman hospitalized for acute lower gastrointestinal hemorrhage. In her medical history, she was previously diagnosed with hypertension, type 2 diabetes mellitus, hypercholesterolemia and ischemic cardiopathy with a reduced ejection fraction to 40%. Her medication consisted of aspirin, bisoprolol, digoxin, lisinopril, molsidomine, gliclazide and pregabalin. She had two kids and was not known for previous blood transfusions.
In the emergency department, her complete blood count showed normocytic anemia with a hemoglobin level of 7.6 g/dL with a mean corpuscular volume of 95 µm3, 267,000 platelets/mm3 and 18,800 white blood cells/mm3, predominantly neutrophils. The absolute reticulocyte count was 161,000/mm3 with a low reticulocyte production index 1.7. Iron studies were below normal values with a ferritin of 30 µg/L with a transferrin saturation of 10%. The patient received two units of cross-matched packed RBCs on day 1 of admission. The patient was transfused again five units total on day 2 and day 5 for recurrent bleeding and an emergent colonoscopy showed diverticulosis.
She was treated with a right colectomy for uncontrolled bleeding and hemodynamic instability on day 4 of her hospitalization. Seven days after the first transfusion, the patient presented with jaundice and a drop of the hemoglobin level to 6.9 g/dL. Hemolytic anemia was diagnosed as haptoglobin levels became undetectable, LDH and total bilirubin rose to 467 U/L (normal range: 135 - 214) and 9.5 mg/dL, respectively. The patient did not show any clinical sign of bleeding. Angiography and abdominal computed tomography ruled out any recurrence of bleeding. Since her hemoglobin dropped lower than the level pre-transfusion, DHTR with HS was suspected (Fig. 1).
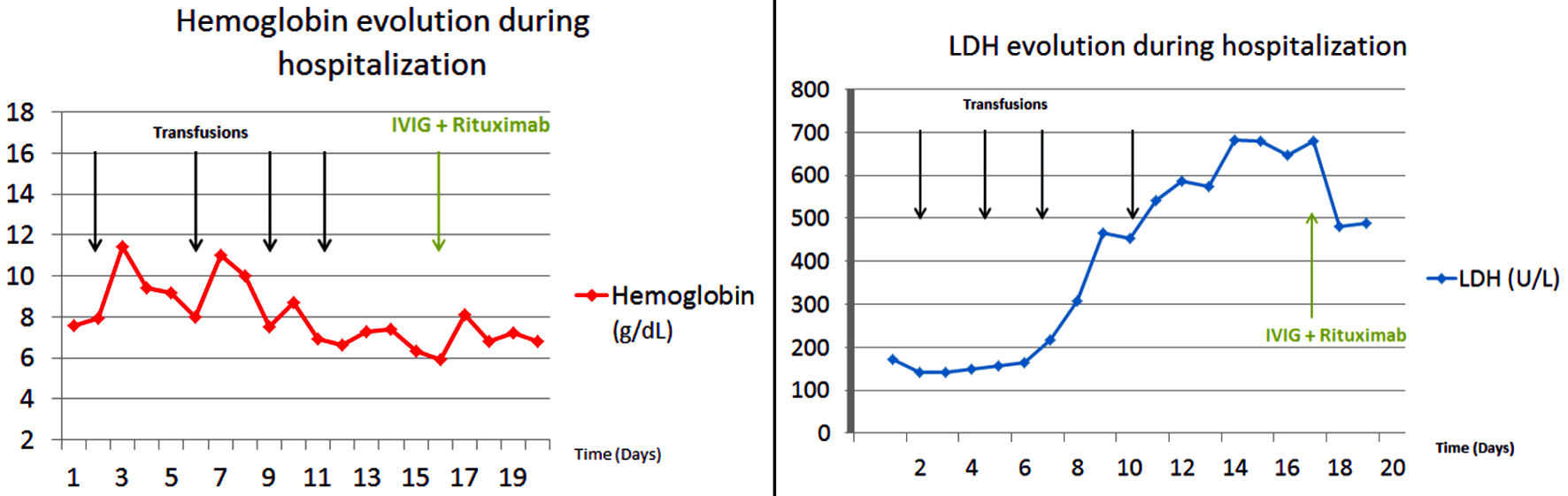 Click for large image | Figure 1. Hemoglobin and LDH evolution during hospitalization. LDH: lactate dehydrogenase; IVIG: intravenous immunoglobulins. |
Diagnosis
The direct antiglobulin test (DAT, Coombs test) was 4+ positive for immunoglobulin G (IgG) and C3b. Anti-C, anti-Jkb and anti-Fya alloantibodies were detected. Her erythrocyte genotype (Table 1) performed after her DHTR confirmed she was Jkb-negative and Fya-negative. The extended blood phenotype of two of the units transfused was C+ c+ K- and C- c+ K+ k+, explaining the incompatibility reaction and suggesting the hemoglobin drop was not due to active bleeding.
 Click to view | Table 1. Erythrocyte Genotype of the Patient |
Treatment
The patient was treated on day 17 with intravenous immunoglobulins (IVIG) 0.5 g/kg/day for 4 days and injection of epoetin alfa 40,000 IU. She also received anti-CD20 monoclonal antibody rituximab 1 g as alloimmunization prophylaxis for potential further transfusions.
Follow-up and outcomes
At day 18, her hemoglobin level stabilized and LDH started to drop. The patient did not show any recurrence of bleeding nor hemolysis, and was safely discharged from the hospital.
| Discussion | ▴Top |
DHTR is a well-known complication in transfusion medicine especially in SCD. Clinically, DHTR can range from mild symptoms to lethal multi-organ failure, but most patients present symptoms such as jaundice, hemoglobinuria, and fever.
HS is a rare and dreaded complication and needs to be better recognized as its outcome can be fatal. The DAT is frequently negative and reticulocytopenia is often observed [6]. As it remains exceptional, its precise incidence and mortality is not clear and most of the data concern SCD patients.
However, both DHTR and HS have been reported in patients with other underlying hematologic disorders such as thalassemia [7], myelofibrosis [8], anemia of chronic disease [9] and lymphoma [10], possibly as they also need multiple transfusions in the setting of the disease itself or the treatment-induced cytopenias. For example, a patient with non-Hodgkin lymphoma developed a DHTR with the same alloantibody, anti-Jkb, as our patient did, and was successfully treated with steroids and extended matched Jkb-negative transfusions [10].
Furthermore, HS is exceptional outside any hematologic condition. To the best of our knowledge, our case is one of the few described in the literature with HS in a person without any hematologic disorder [11-14].
In HS, the hemoglobin drop below pre-transfusion levels reflects the destruction of both autologous and transfused RBCs. This phenomenon is called “bystander hemolysis”, as the autologous RBCs are destroyed even if they do not present the targeted antigen [6]. The exact mechanism of HS is still poorly understood, but different hypotheses have been suggested, including macrophage activation, suppression of erythropoiesis and complement activation [15].
Alloantibody identification is important for the diagnosis of DHTR and HS. In addition to anti-Jkb, our patient developed also anti-C and anti-Fya alloantibodies which are classically known to be harmful alloantibodies and comforted the alloimmunization suspicion.
Nevertheless, identifying the responsible antibody remains a challenge because of their transient nature. Indeed, due to antibody evanescence, one-thirds of DHTR cases will not have any identified antibody, putting at risk patients for other hemolysis reactions, acute or delayed, if transfusion is again needed [16]. In a study on 124 SCD patients, the most frequent antibodies identified were Rh antibodies (anti-D, anti-C, anti-E, and anti-e) [2]. Anti-Jkb was less frequent than anti-Jka in a study in India on 405 alloantibodies; and in 32% of cases, multiple alloantibodies were involved [17].
Alloimmunization should always be kept in mind in front of an unexplained post-transfusion hemolysis, even in patients without transfusion history. Indeed, in addition to our patient case description, few cases have been reported in the literature. A patient without previous transfusions suffered from a fatal DHTR due to Kell alloantibodies thought to be related to a medical history of 13 pregnancies [11]. An previous report of a 58-year-old patient from Haiti who had never been transfused in the past but was known with a history of five pregnancies and two abortions, presented with a severe DHTR due to anti-U alloimmunization after being transfused for rectal bleeding [18]; reminding that transfusions are not the only way to get alloimmunized, and obstetric history needs to be thorough.
Massive transfusions in hemorrhagic shocks are also a potential alloimmunization risk. Similar to our patient who was acutely transfused for uncontrolled gastrointestinal bleeding, a young multi-transfused patient known with prehepatic portal hypertension and bleeding varices, presented a HS successfully treated with corticosteroids and immunoglobulins [14]. Also in the context of massive transfusions, Eberly et al reported the case of a 55-year-old patient transfused 10 units of RBCs after a motor vehicle accident who presented DHTR/HS with Jka alloantibodies and was not known for a hematologic disease [13]. Finally, intravenous drug abuse can be another potential cause for alloimmunization as Lappen et al showed in their study on pregnant women [19].
Given the complexity and rarity of HS, management should always be decided after discussion with the hematologist and transfusion medicine specialist. Avoidance of further transfusions is of major importance unless the patient has life-threatening anemia. First-line treatment after avoidance of transfusions is IVIG for 3 to 5 days and high-dose corticosteroids as recommended by the 2020 guidelines of the American Hematology Society [20]. High-dose erythropoietin should be given if reticulocytopenia is present and supportive care with intravenous iron should also be considered. In severe HS with organ damage, eculizumab, an anti-C5 monoclonal antibody, can be administered given that complement activation is suspected to contribute to HS although the outcome is variable [16, 21].
Recently, a life-threatening HS in a SCD patient has been successfully treated with tocilizumab, an anti-interleukin (IL)6 monoclonal antibody, suggesting macrophage activation might play a role in its mechanism and could be a potential therapeutic target [17].
These patients need extended-matching RBCs and to be monitored closely after transfusions. In SCD patients, HbA repeated dosage should be followed to exclude transfused RBCs hemolysis and the use of a diagnostic nomogram can be a helpful tool in the process [18]. A systematic antibody screening post transfusion and a thorough transfusion and alloimmunization history need to be kept in patient’s medical files to bring awareness to this dangerous condition, and a nationwide antibody database could help prevent this complication [22]. Genotyping blood group antigen should be done when possible as it allows genotype-compatible transfusions in the future [5]. If transfusion is deemed necessary, an extended-matching of RBC needs to be done to identify new alloantibodies (C/c, E/e, K, Jka/Jkb, S/s, M/N, Fya/Fyb,) [20]. Prophylaxis with steroids and/or rituximab prior to the transfusion can lower the risk of additional alloimmunization and delayed reaction in high-risk patients who might need future transfusions [20, 21].
Learning points
Still underestimated even within the SCD population, HS is a potentially life-threatening condition which can also affect individuals without an underlying hematologic condition. Despite all matching safety measures, DHTR and HS need to be suspected by clinicians even in non-SCD patients, as a prompt recognition will stop additional transfusions worsening the hemolysis and allow for adequate treatment.
Acknowledgments
None to declare.
Financial Disclosure
The authors did not receive any financial support for the manuscript.
Conflict of Interest
The authors deny any conflict of interest.
Informed Consent
Verbal informed consent was obtained from the patient.
Author Contributions
KE did the literacy search. KE, FSB and MC all contributed equally to the writing of the manuscript and to the figures.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
DHTR: delayed hemolytic transfusion reaction; DAT: direct antiglobulin test; HS: hyperhemolysis syndrome; LDH: lactate dehydrogenase; RBC: red blood cell; SCD: sickle cell disease
| References | ▴Top |
- Coleman S, Westhoff CM, Friedman DF, Chou ST. Alloimmunization in patients with sickle cell disease and underrecognition of accompanying delayed hemolytic transfusion reactions. Transfusion. 2019;59(7):2282-2291.
doi pubmed - Habibi A, Mekontso-Dessap A, Guillaud C, Michel M, Razazi K, Khellaf M, Chami B, et al. Delayed hemolytic transfusion reaction in adult sickle-cell disease: presentations, outcomes, and treatments of 99 referral center episodes. Am J Hematol. 2016;91(10):989-994.
doi pubmed - Vichinsky EP, Earles A, Johnson RA, Hoag MS, Williams A, Lubin B. Alloimmunization in sickle cell anemia and transfusion of racially unmatched blood. N Engl J Med. 1990;322(23):1617-1621.
doi pubmed - Banks M, Shikle J. Hyperhemolysis syndrome in patients with sickle cell disease. Arch Pathol Lab Med. 2018;142(11):1425-1427.
doi pubmed - Narbey D, Habibi A, Chadebech P, Mekontso-Dessap A, Khellaf M, Lelievre JD, Godeau B, et al. Incidence and predictive score for delayed hemolytic transfusion reaction in adult patients with sickle cell disease. Am J Hematol. 2017;92(12):1340-1348.
doi pubmed - Treleaven JG, Win N. Hyperhaemolysis syndrome in a patient with myelofibrosis. Hematology. 2004;9(2):147-149.
doi pubmed - Hussain SS, Ebbs AM, Curtin NJ, Keidan AJ. Delayed haemolytic transfusion reaction due to anti-Jkb in a patient with non-Hodgkin's lymphoma-transient nature of anti-Jkb and the importance of early serological diagnosis. Transfus Med. 2007;17(3):197-199.
doi pubmed - Merrill SA, Brodsky RA, Lanzkron SM, Naik R. A case-control analysis of hyperhemolysis syndrome in adults and laboratory correlates of complement involvement. Transfusion. 2019;59(10):3129-3139.
doi pubmed - Fasano RM, Miller MJ, Chonat S, Stowell SR. Clinical presentation of delayed hemolytic transfusion reactions and hyperhemolysis in sickle cell disease. Transfus Clin Biol. 2019;26(2):94-98.
doi pubmed - Makroo RN, Nayak S, Chowdhry M, Karna P. Facts and fallacies of kidd antibodies: experience in a tertiary care hospital in North India. Indian J Hematol Blood Transfus. 2017;33(2):254-258.
doi pubmed - Taliano V, Fleury M, Pichette R, Lamothe M, Decary F. [Delayed hemolytic transfusion reaction caused by an anti-U]. Rev Fr Transfus Hemobiol. 1989;32(1):17-26.
doi - Chubar E, Bisharat N. Fatal delayed haemolytic transfusion reaction in a patient without previous transfusions but with an obstetric history of 13 pregnancies. BMJ Case Rep. 2017;2017:bcr-2017-222343.
doi pubmed - Eberly LA, Osman D, Collins NP. Hyperhemolysis Syndrome without Underlying Hematologic Disease. Case Rep Hematol. 2015;2015:180526.
doi pubmed - Chou ST, Alsawas M, Fasano RM, Field JJ, Hendrickson JE, Howard J, Kameka M, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020;4(2):327-355.
doi pubmed - Gupta S, Fenves A, Nance ST, Sykes DB, Dzik WS. Hyperhemolysis syndrome in a patient without a hemoglobinopathy, unresponsive to treatment with eculizumab. Transfusion. 2015;55(3):623-628.
doi pubmed - Dumas G, Habibi A, Onimus T, Merle JC, Razazi K, Mekontso Dessap A, Galacteros F, et al. Eculizumab salvage therapy for delayed hemolysis transfusion reaction in sickle cell disease patients. Blood. 2016;127(8):1062-1064.
doi pubmed - Lee LE, Beeler BW, Graham BC, Cap AP, Win N, Chen F. Posttransfusion hyperhemolysis is arrested by targeting macrophage activation with novel use of Tocilizumab. Transfusion. 2020;60(1):30-35.
doi pubmed - Mekontso Dessap A, Pirenne F, Razazi K, Moutereau S, Abid S, Brun-Buisson C, Maitre B, et al. A diagnostic nomogram for delayed hemolytic transfusion reaction in sickle cell disease. Am J Hematol. 2016;91(12):1181-1184.
doi pubmed - Lappen JR, Stark S, Gibson KS, Prasad M, Bailit JL. Intravenous drug use is associated with alloimmunization in pregnancy. Am J Obstet Gynecol. 2016;215(3):344 e341-346.
doi pubmed - Pirenne F, Yazdanbakhsh K. How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions. Blood. 2018;131(25):2773-2781.
doi pubmed - Noizat-Pirenne F, Bachir D, Chadebech P, Michel M, Plonquet A, Lecron JC, Galacteros F, et al. Rituximab for prevention of delayed hemolytic transfusion reaction in sickle cell disease. Haematologica. 2007;92(12):e132-135.
doi pubmed - Powell Z, Jiang N, Shrestha R, Jackson DE. Would a National Antibody Register contribute to improving patient outcomes? Blood Transfus. 2021;20(2):132-142.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.