

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 11, Number 1, February 2022, pages 21-28

Concurrent Presentation of Hairy Cell Leukemia and Mantle Cell Lymphoma (Leukemic Non-Nodal Variant): An Extremely Rare Composite Lymphoma
Dina S. Solimana, b, d, e ![]() , Feryal Ibrahima, Liam J. Fernyhoughb, Farzana Muradc, Susanna Akikic
, Feryal Ibrahima, Liam J. Fernyhoughb, Farzana Muradc, Susanna Akikic
aDepartment of Laboratory Medicine and Pathology, National Center for Cancer Care and Research, Hamad Medical Corporation, Doha, Qatar
bWeill Cornell Medicine-Qatar, Doha, Qatar
cDiagnostic Genomic Division, Hamad Medical Corporation (HMC), Doha, Qatar
dDepartment of Clinical Pathology, National Cancer Institute, Cairo, Egypt
eCorresponding Author: Dina Sameh Soliman, Department of Laboratory Medicine and Pathology, Hamad Medical Corporation, Doha, Qatar
Manuscript submitted October 23, 2021, accepted December 18, 2021, published online January 10, 2022
Short title: Concurrent HCL and L-NN-MCL
doi: https://doi.org/10.14740/jh942
| Abstract | ▴Top |
Herein, we describe the clinicopathologic and genetic characteristics of the first report of simultaneous bone marrow involvement by classical hairy cell leukemia (HCL) and leukemic non-nodal variant of mantle cell lymphoma (L-NN-MCL) with t(11;14)(q13;q32) with BRAF mutation and deletion of TP53. A 40-year-old asymptomatic man was investigated for incidental neutropenia and thrombocytopenia. Flow cytometry showed two distinct monotypic B-cell populations: one expressed CD19 (bright), CD20 (bright), FMC7, CD103, CD25, CD11c, CD123, and IgD (bright) and showed kappa light chain restriction (bright), consistent with HCL and the other kappa-restricted CD5/CD10-negative B-cell population with distinctive immunophenotypic features. The bone marrow biopsy is infiltrated by an abnormal B-lymphoid infiltrate with different patterns of infiltration in different marrow areas. Fluorescence in situ hybridization (FISH) analysis revealed a CCND1/IGH rearrangement, t(11;14)(q13;q32), and deletion of TP53. The BRAF V600E missense mutation was detected by quantitative real-time polymerase chain reaction (PCR). The diagnosis of a composite B-cell neoplasm was composed of HCL together with a second CD5/CD10-negative monotypic B-cell population, with CCND1/IGH fusion, favoring the 2016 WHO new category of L-NN-MCL (CD5/SOX11-negative). Treatment with cladribine and rituximab normalized the blood counts within 6 weeks without significant side effects. L-NN-MCL is one of the smoldering MCL subtypes, recently listed in WHO 2016 as a separate variant, with a particular set of unique features and a less aggressive clinical course compared to classical MCL. To date, the clinicopathological features (including the bone marrow findings) of L-NN-MCL have not been sufficiently characterized in the literature. We describe the first report of synchronous presentation of HCL and L-NN-MCL. This case represents a real challenge from the biologic, diagnostic and therapeutic point of views, due to extremely rare combination of two distinct uncommon B-cell neoplasms. The study of composite lymphomas offers the opportunity to evaluate the etiology and the clonal interrelationship involved in the pathogenesis/evolution of lymphomas.
Keywords: Hairy cell leukemia; Leukemic non-nodal mantle cell lymphoma; Composite lymphoma
| Introduction | ▴Top |
Composite lymphoma (CL), defined as two or more distinctly different lymphomas occurring concurrently at the same organ or tissue at the time of presentation, is a rare event with estimated incidence at 1-4% of lymphoma presentations [1]. Hairy cell leukemia (HCL) is an uncommon but distinct B-cell lymphoid neoplasm [2]. Morphologically, hairy cells (HCs) are characteristically small- to medium-sized with oval or indented nuclei, an abundant cytoplasm and typical hairy-like projections. HCL has a unique immunophenotype (IPT) characterized by clonal expansion of B cells with bright CD19, CD20, CD22, and CD200 expression. HCs are usually negative for CD5, CD23, and CD10 and characteristically positive for CD11c, CD103, CD123, and CD25.
Underpinning the characteristic morphologic and phenotypic features of classical HCL are activating mutations in the kinase domain of the BRAF gene. The somatic V600E BRAF mutation in exon 15 is present in almost 100% of classical HCLs [3] and is now considered the molecular hallmark of HCL and an early genetic event that can be used as a novel diagnostic tool and an opportunity for therapies targeting of BRAF (BRAF inhibitors) [4]. The late-activated post-germinal center memory B cell is considered as the cell of origin for HCL [5].
On the other hand, mantle cell lymphoma (MCL) is another B-cell malignancy that makes up approximately 6% of all the non-Hodgkin lymphomas [6]. MCL is derived from naive, pre-germinal center cells of primary follicles or the mantle zone of secondary follicles. It presents most commonly with generalized lymphadenopathy, hepatosplenomegaly, and extra nodal disease which primarily involves the bone marrow (BM) and gastrointestinal tract [2]. MCL typically possesses the characteristic t(11;14)(q13;q32) chromosomal translocation leading to overexpression of cyclin-D1 as the chief genetic event, and provides an exceptional marker for diagnosis. MCL cells conventionally express CD5 and cyclin-D1 while lacking CD10, CD23, and BCL-6 expression.
Traditionally, MCL has been considered to have a more aggressive clinical course with shorter survival (median survival of 3 - 5 years) [2, 7], compared with other indolent lymphomas (including HCL).
Recent data show that MCL is in fact a heterogenous entity with up to 30% of MCL patients having a more indolent clinical course and proliferation activity [8], with survival exceeding 7 - 10 years referred to as smoldering MCL [9].
Various clinicopathologic variables aid in the stratification of patients with MCL; these include baseline clinical features Mantle Cell International Prognostic Index (MIPI), leukemic non-nodal and in situ presentation, pathological features including blastoid morphology, Ki-67 proliferation index, SOX11 expression, and genetic aspects (immunoglobulin mutation status, TP53, and CDKN2A deletion) [10]. Leukemic non-nodal MCL (L-NN-MCL) is one of the smoldering MCL subtypes, recently listed in WHO 2016 as a separate variant, with a particular set of unique features and a less aggressive clinical course compared to classical MCL [2, 11]. To date, the clinicopathological features (including the BM findings) of L-NN-MCL have not been sufficiently characterized in the literature.
We describe the first report of synchronous presentation of HCL and L-NN-MCL. This case represents a real challenge from the biologic, diagnostic and therapeutic point of views, due to extremely rare combination of two distinct uncommon B-cell neoplasms.
| Case Report | ▴Top |
Investigations
A 40-year-old man, previously healthy and asymptomatic, was referred to hematology clinic after the incidental finding of thrombocytopenia and neutropenia in his routine laboratory workup. The patient denied any fever, weight loss, night sweats or bleeding tendency. He had a previous history of lymphadenopathy 4 years previously, when a lymph node (LN) biopsy revealed reactive follicular hyperplasia.
Initial investigations revealed low-normal white blood cells at 4.4 × 103/µL (n: 4 - 10), mild neutropenia (absolute neutrophil count at 1.3 × 103/µL), moderate thrombocytopenia (platelets: 77 × 103/µL; n: 150 - 400) and within normal hemoglobin level. His initial workup showed normal liver and kidney function tests with normal lactate dehydrogenase (LDH). Abdominal ultrasound revealed mild splenomegaly (13 cm).
A fluorodeoxyglucose (FDG) positron emission tomography/computed tomography (PET/CT) scan showed an enlarged spleen (15 cm) with intense FDG uptake with standardized uptake value (SUV) max of 5.6 compared to liver (2.1). A small (1.5 cm) axillary LN with barely appreciated tracer uptake (SUV max of 2.2) was detected, probably reactive.
Diagnosis
Peripheral blood (PB) smear (Fig. 1a) showed moderate neutropenia and thrombocytopenia with about 7% abnormal lymphoid cells (HCs) of small/medium to large size with oval/indented/kidney shaped nuclei, dispersed nuclear chromatin, few small nucleoli with abundant pale cytoplasm and hairy-like projections in some forms.
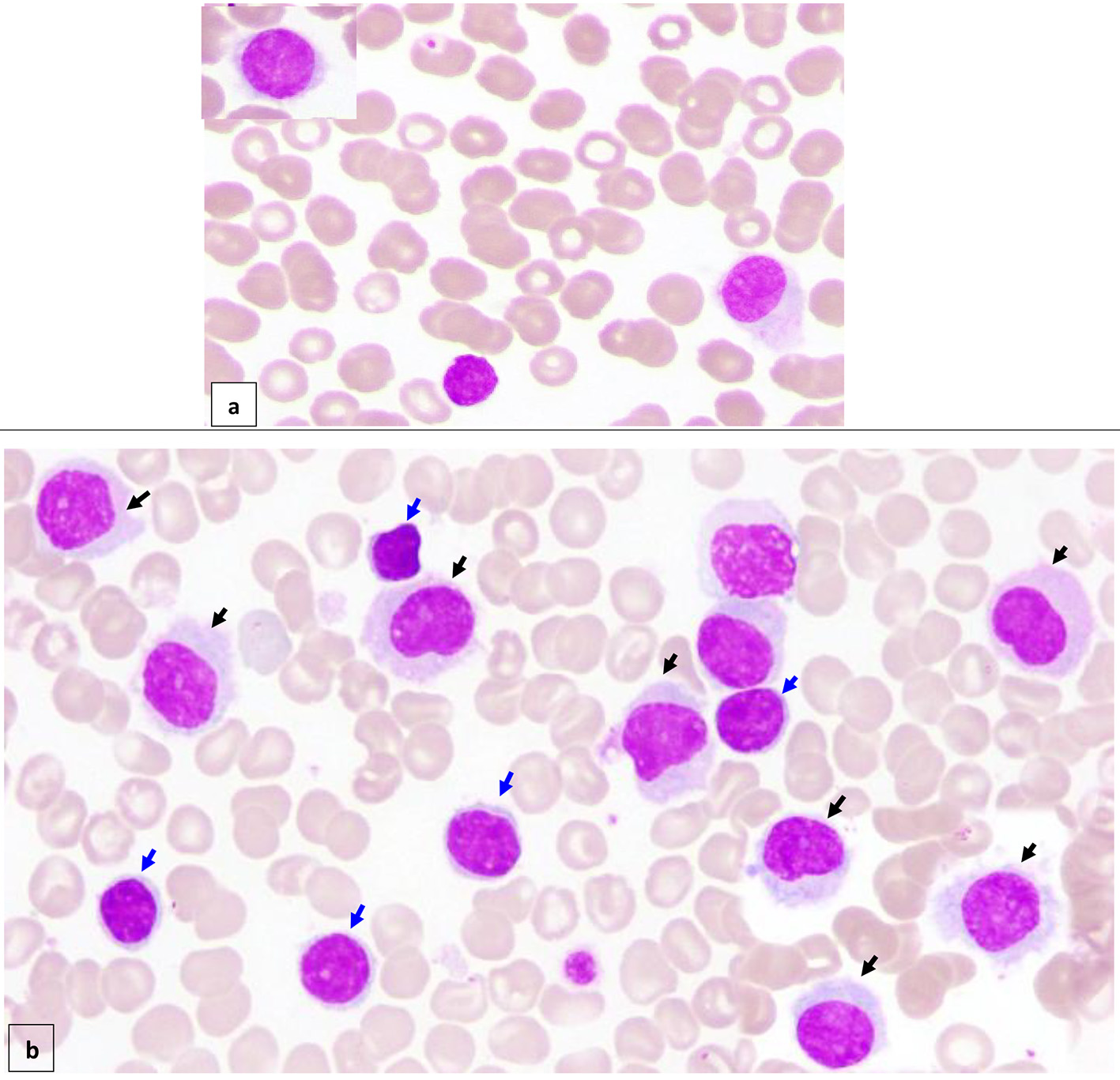 Click for large image | Figure 1. Peripheral blood (× 100) shows about 7% circulating abnormal lymphoid cells (hairy cells) of small/medium to large size with oval/indented/kidney shaped nuclei, dispersed nuclear chromatin, few small nucleoli, abundant pale cytoplasm and hairy like projections in some forms (a) (black arrow). Bone marrow aspirate (× 100) is infiltrated by about 16% abnormal lymphoid cells (hairy cells) (black arrow). In addition, there are small mature looking lymphocytes (about 18%) with condensed nuclear chromatin a small rim of cytoplasm (b) (blue arrow). |
BM aspirate (Fig. 1b) was infiltrated by about 16% abnormal lymphoid cells (HCs). In addition, there were some mature looking lymphocytes (about 18%) including plasmacytoid forms and a few forms with irregular cytoplasmic projections.
Flow cytometry (FCM) (on BM aspirate) (Fig. 2) showed a population of monotypic B cells comprising about 6% showing high light side scatter and expressing CD19 (bright), CD20 (bright), CD79b, FMC7, CD103, CD25, CD11c, CD123, IgM, and IgD (bright) and showing kappa light chain restriction (bright). This monotypic population was negative for CD5 and CD10 with no significant expression of CD38, CD23 or CD43. This IPT is consistent with HCL.
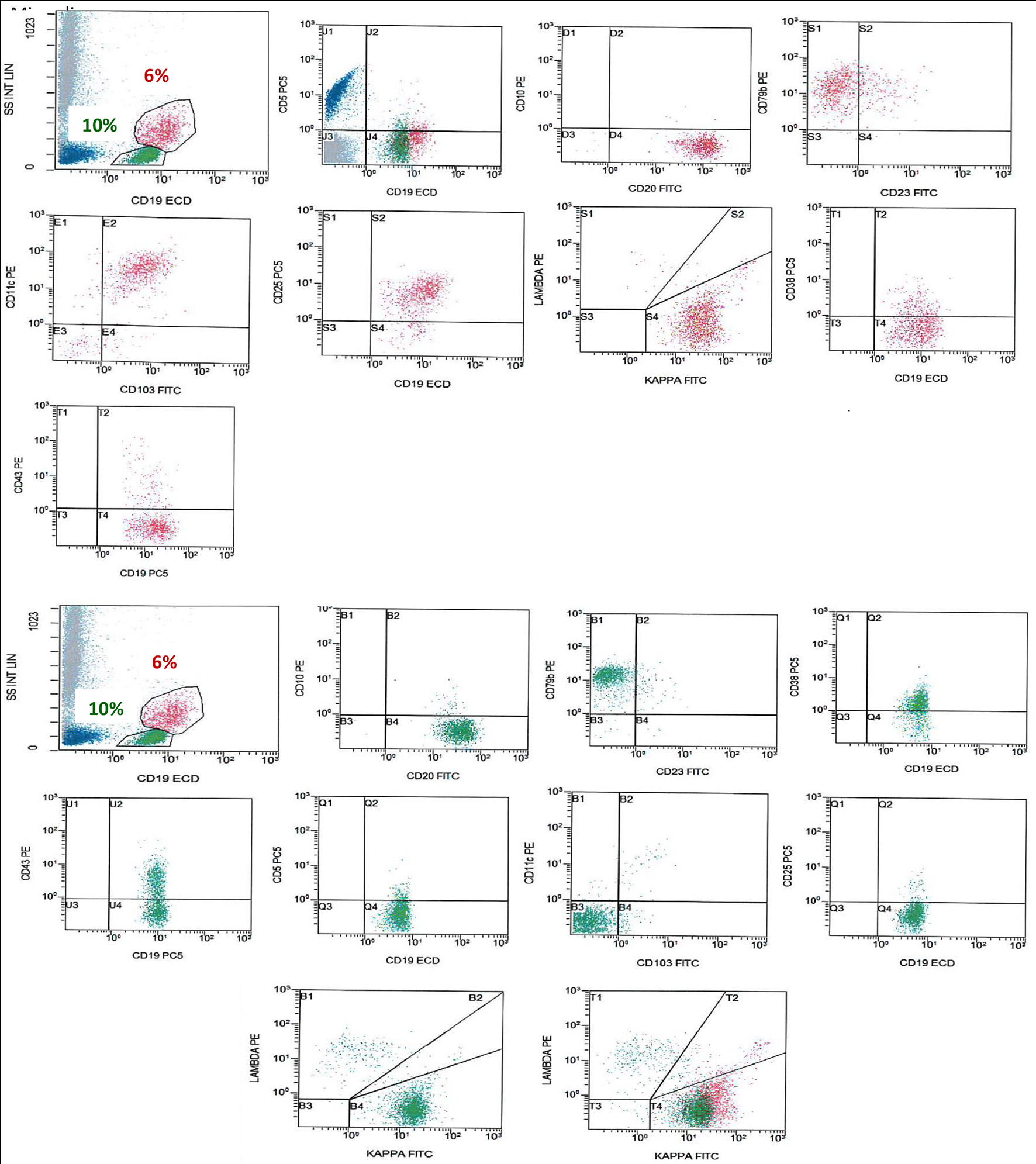 Click for large image | Figure 2. FCM on BM shows a population of monotypic B cells (red), showing high light side scatter and expressing CD19 (bright), CD20 (bright), CD79b, CD103, CD25, CD11c, and shows kappa light chain restriction (bright). This monotypic population is negative for CD5, CD10 and CD43, an immunophenotype consistent with HCL. There is another population of monotypic B cells (green), showing lower light scatter and expressing CD19, CD20, CD79b, and CD38 with partial expression of CD43 and shows kappa light chain restriction. This population is negative for CD5, CD23, CD10, CD25, CD11c and CD103. FCM: flow cytometry; BM: bone marrow; HCL: hairy cell leukemia. |
In addition, the analysis revealed a second population of kappa-restricted monotypic B cells (about 10%), showing lower light scatter and expressing CD19, CD20, FMC7, CD79b, IgM, IgD and CD38 with partial expression of CD43 (heterogenous). This population was negative for CD5, CD23, CD10, CD25, CD11c, CD103 and CD123.
BM biopsy (BMB) (Fig. 3) showed variable cellularity ranging between marked hypocellularity (< 5-20%) cellularity in few spaces alternating with normocellular and mildly hypercellular areas (70-85%).
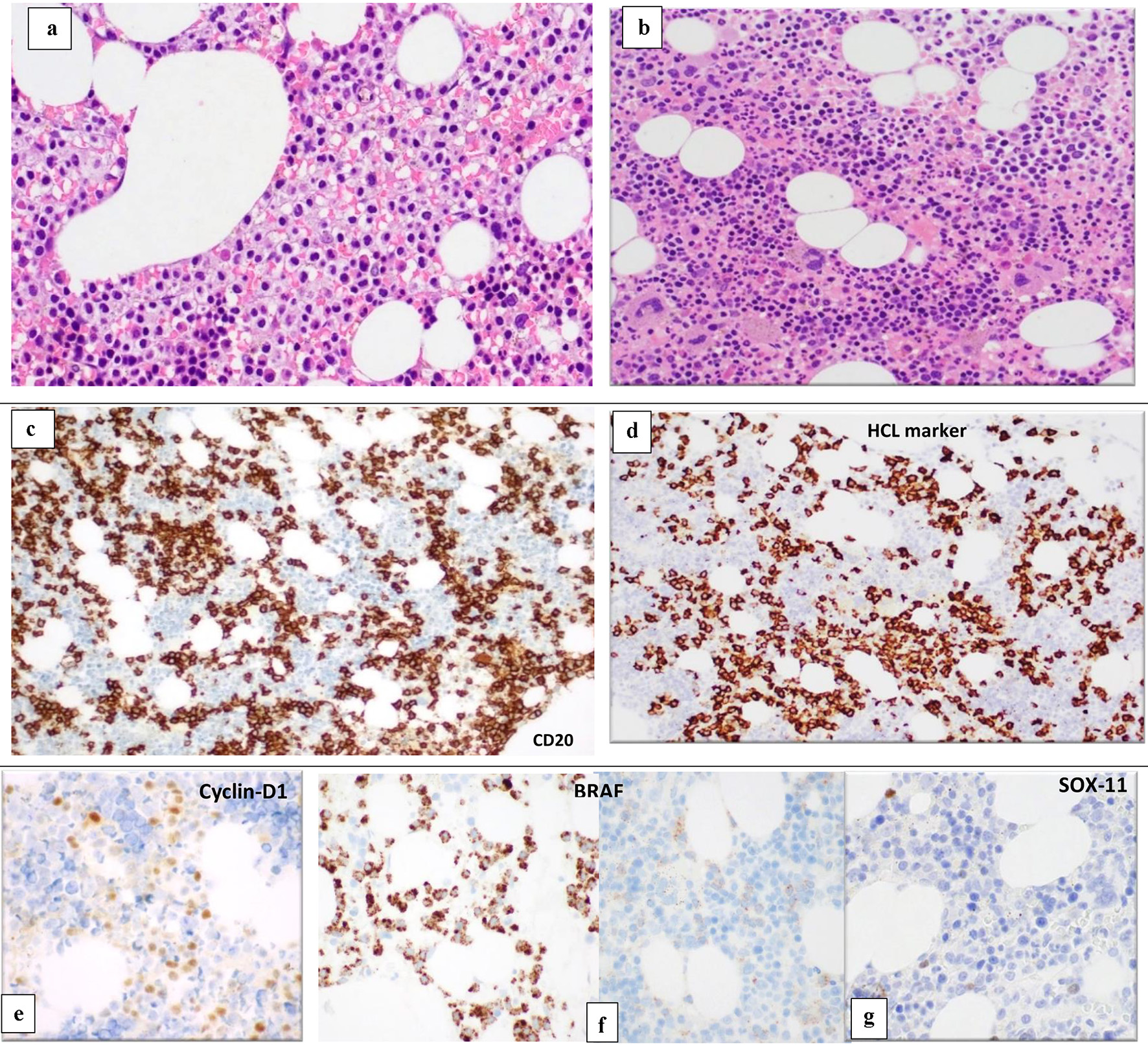 Click for large image | Figure 3. BM biopsy (H&E, × 10) shows variable cellularity with a lymphoid infiltrate showing different patterns of infiltration in different marrow areas. Some areas show widely spaced infiltrate composed of small- to medium-sized lymphoid cells with kidney shaped/indented nuclei and abundant cytoplasm giving the fried-egg appearance (a). Other BM spaces show an interstitial infiltration with small sized lymphoid cells (b). The abnormal lymphoid cells are positive for CD20 (c) and show partial positivity for DBA-44 (HCL marker) (d), cyclin-D1 (e), BRAF (positive in areas with HCL infiltrate) (f) and negative for SOX11 (g). BM: bone marrow; H&E: hematoxylin and eosin; HCL: hairy cell leukemia. |
The BM was infiltrated with many B-lymphoid cells, but with different patterns of infiltration in different marrow areas. Some areas show widely spaced infiltrate composed of small- to medium-sized lymphoid cells with kidney shaped/indented nuclei and abundant cytoplasm giving the fried-egg appearance. Other BM spaces showed an interstitial infiltration with small-sized lymphoid cells. The abnormal lymphoid cells are positive for CD20, PAX-5, and showed partial positivity for DBA-44, cyclin-D1, CD25 and annexin A1. Positivity for CD20 and PAX5 exceeded that for DBA44, cyclin-D1 and CD25. No increase in plasma cells was highlighted with CD138 immunostain.
The fluorescence in situ hybridization (FISH) analysis (Fig. 4a, b) on BM revealed an abnormal hybridization signal pattern consistent with CCND1/IGH rearrangement, t(11;14)(q13;q32) and deletion of TP53 in 7% of the cells analyzed.
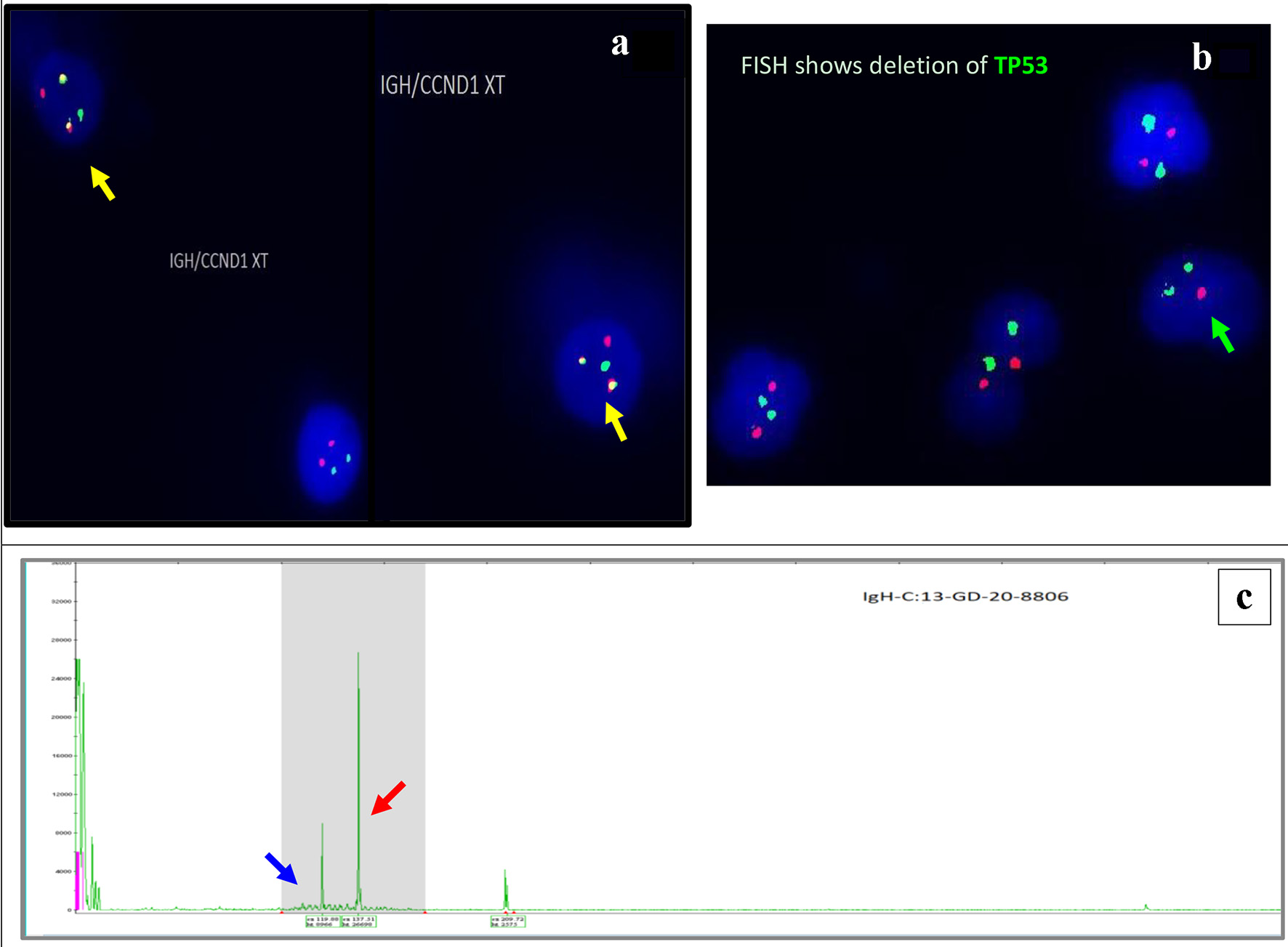 Click for large image | Figure 4. Interphase FISH demonstrating IGH/CCND1 rearrangement (a): IGH (14q32) labelled with spectrum green, CCND1 (11q13) labelled with spectrum orange show dual fusions signals as result of IGH/CCND1 fusion (yellow arrow). Loss of a single copy of CEP17/TP53 indicates deletion of TP53 gene on chromosome 17. CEP17 (17p11.1-q11.1) labelled with spectrum green, and TP53 (17p13.1) labelled with spectrum orange (b) (green arrow). PCR-based clonality testing (Invivoscribe Identiclone) was used to identify clonal B-cell populations with primers that target conserved framework (FR) and joining (J) regions of the IGVH genes. Two clonal B-cell populations were identified: a dominant 335 bp IGH VH (FR1) JH clone (red arrow) and a 317 bp IGH VH (FR1) JH subclone (blue arrow) (c). FISH: fluorescence in situ hybridization; PCR: polymerase chain reaction. |
Because of its challenging nature, the whole diagnostic material was referred abroad to Mayo Clinic reference laboratories for second opinion, where they performed immunohistochemistry for BRAF and repeated cyclin-D1 immunostain, and confirmed the diagnosis of a composite B-cell neoplasm composed of HCL together with a second monotypic B-cell population, CD5/CD10-negative with CCND1/IGH fusion by FISH, favoring the 2016 WHO new category of L-NN-MCL.
A low level positive BRAF V600E missense mutation was detected by quantitative real-time polymerase chain reaction (PCR) confirming the presence of a minor HCL clone.
Immunoglobulin heavy chain variable region gene rearrangement analysis showed two distinct clonal B-cell populations, a dominant 335 bp IGVH (FR1) clone and a 317 bp IGVH (FR1) subclone (Fig. 4c).
Taken together, the genetic results support the presence of two clonal B-cell populations, a dominant clone with a TP53 deletion and CCND1/IGH rearrangement and a second BRAF V600E mutated B-cell subclone.
Treatment
The multidisciplinary leukemia team supported the approach of giving cladribine first to treat the HCL component and then to reassess the mantle cell component afterwards.
Initial treatment was aimed at the HC component and consisted of cladribine 0.14 mg/kg intravenous (IV) daily for 5 days and rituximab 375 mg/m2 IV weekly for eight doses.
Follow-up and outcomes
The thrombocytopenia and neutropenia resolved within 6 weeks of starting treatment without significant side effects and the patient achieved end of treatment complete remission confirmed by PET/CT and BMB.
| Discussion | ▴Top |
HCL is an uncommon, chronic B-cell leukemia, first reported as a distinct entity in the 1950s [1, 2]. HCL accounts for 2% of lymphoid leukemias, with a male predominance and median age at diagnosis of 58 years. Classical HCL and its variant form (HCL-V) are now regarded as separate entities [3], with different cytological, hematological and immunophenotypic features. BRAF V600E mutation, present in virtually 100% of cases of classical HCL [4], is regarded as a disease-defining event.
This case represents a rare model of CL combining two distinct B-cell neoplasms (HCL and MCL) and the first reported description of HCL and L-NN-MCL. The divergent morphology, different histopathologic infiltration pattern and different immunophenotypic features (by FCM and immunohistochemistry (IHC)), confirmed by molecular genetics findings, all supported the presence of two distinctive types of lymphomas.
Although both clones were kappa-restricted, the FCM immunophenotyping had clearly separated them based on different cell characteristics (size and complexity) and distinct IPT (HCL and non-HCL clones). The detection of CCND1/IGH fusion by FISH and a second immunophenotypically distinct, clonal B-cell population by FCM and given that CCND1-IGH fusion is rarely in HCL, the upregulation of cyclin-D1 expression detected by IHC stems from a different pathophysiologic process, and this would support the presence of a second separate clonal process. With this information, based on FCM IPT, the population, harboring the CCNDA-IGH fusion is probably the (non-HCL) CD5/CD10-negative population by FCM. The presence of the translocation would then support a diagnosis of MCL, probably L-NN-MCL.
The differential diagnosis of CD5/CD10-negative lymphomas would classically include marginal zone lymphoma and lymphoplasmacytic lymphoma, among other mature B-cell neoplasms that may present with an atypical IPT. However, no pathologic, genetic or clinical evidence for any of the mentioned categories was found.
In both neoplasms, the main tumor burden was localized in BM (and likely the spleen), whereas the LNs were minimally (if at all) involved.
Clinically L-NN-MCL involves the PB, BM and sometimes the spleen. However, this variant is characterized by reduced cell adhesion/infiltrative properties and hence lacks significant lymphadenopathy (defined as peripheral LNs < 1 - 2 cm and absent lymphadenopathy on CT) [12-14]. A thorough radiological staging CT or, when available, PET/CT scan to demonstrate the absence of significant lymphadenopathy is required for the diagnosis of L-NN-MCL variant.
Morphologically, the L-NN-MCL leukemic cells are small, resembling chronic lymphocytic leukemia (CLL) type cells with no specific morphologic features. L-NN-MCL tends to present with a lower MIPI/Ki-67 proliferation rate and harbors somatic Ig hypermutation [14], demonstrating their generally less aggressive features. CD5 negativity is more frequent than in classic MCL [1]. In addition, recent published data also show decreased SOX11 expression in patients with indolent (smoldering) MCL including L-NN-MCL [12-15].
Patients with L-NN-MCL tend to have a more favorable outcome than those with classic MCL (median survival: 79 months). A “watch and wait” approach or initially less intensive treatment options can be successful for long periods [12-14].
Some cases of L-NN-MCL show progressive disease with or without tissue involvement, and/or transformation to a morphologically aggressive variant. This evolution is frequently associated with acquisition of TP53 mutations (17p13) or other oncogenic mutations [16].
The case reported here although clinically indolent, has a low level of TP53 mutation detected, this being a worrisome finding that may imply a more aggressive clinical course in the future.
CLs involving HCL are rare with scarce case reports, mostly T-cell lymphomas presenting in association with HCL. The reported cases include associated peripheral T-cell lymphoma [17], hepatosplenic T-cell lymphoma [18], and cutaneous T-cell lymphoma [19]. Among B-cell neoplasms, B-CLL has been the most common subtype reported in association with HCL [20-22].
Synchronous development of HCL and MCL is extremely rare with a single case reported in 2019 [22]. The group demonstrated independent origin of two tumor components and proved the different way of their differentiation. Only a single case of CL involving L-NN-MCL in association with gamma-delta T-large granular lymphocytic leukemia in a patient with rheumatoid arthritis has been reported [23]. No previous reports of HCL with L-NN-MCL have been found.
Among the theories explaining the pathogenesis of CL, we propose that the immune dysregulation and immunodeficiency status commonly associated with HCL seem the most likely to explain the propensity to develop a secondary neoplastic clone (MCL) in our case.
It appears less likely that there has been transformation from one lymphoma type to another in this context as both components (HCL and L-NN-MCL) are considered to be low-grade lymphomas. These two lymphoid malignancies also have different genetic drivers, decreasing the likelihood of the possibility of a common genetic predisposition for both components. Additionally, the presence of completely different genetic drivers (in each lymphoma component) would exclude the possibility of genetic predisposition for both components.
Conclusion
In summary, we describe the first case report of synchronous presentation of HCL and L-NN-MCL. This case represents a real challenge due to extremely rare combination of two separate uncommon B-lymphoproliferative disorders.
These two neoplastic processes have different cells of origin, clinical behavior with distinctive morphologic, immunophenotypic, cytogenetic and molecular genetic characteristics; in particular each of these neoplasms has its own unique genetic signature with (t(11;14) in MCL and the BRAF mutation in HCL. Two distinct clonal B-cell populations were confirmed by PCR analysis of the IGVH gene rearrangements. Further analysis of IGVH gene family usage, although not performed here, could help distinguish if these are two independent clones or if both clones share a common germinal cell origin.
Learning points
The diagnosis of the HCL component in our case was straightforward, given the characteristic morphology, IPT and genetic features. However, the greater diagnostic challenge was related to the diagnosis of L-NN-MCL component which was overshadowed by a number of atypical features including the lack of CD5 and SOX11 expression in addition to the known frequent expression of cyclin-D1 by HCL cells and absence of lymphadenopathy or other tissue involvement, which added another level of complexity in making the diagnosis. There was a convincing evidence for BM involvement by two distinct lymphomas. The pathologists should be aware about the presence of this variant of MCL (with atypical features) that can be easily missed and necessitates an integrated analysis of multiparameteric diagnostic data compiling morphologic, immunophenotypic, cytogenetics and molecular genetics findings.
Acknowledgments
We acknowledge the contributions of the Hematology Team in Clinical Hematology Department particularly Dr. Halima El-Omri and Dr. Amna Gamil and we also would like to acknowledge the role of flow cytometry, cytogenetics and molecular genetics teams in the Department of Laboratory Medicine and Pathology, Hamad Medical Corporation.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
DSS performed the bone marrow examination, literature review and formulated the paper; FI reviewed the diagnosis and the manuscript; LJF reviewed the clinical data, treatment plans and reviewed the manuscript; FM performed the cytogenetics testing; SA performed the molecular genetics testing and reviewed the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
CL: composite lymphoma; HCL: hairy cell leukemia; MCL: mantle cell lymphoma; BM: bone marrow; L-NN-MCL: leukemic non-nodal MCL
| References | ▴Top |
- Gulati R, Zhou J. Composite Lymphoma. In: Wang E, Lagoo A (eds). Practical lymph node and bone marrow pathology. Practical Anatomic Pathology. Springer, Cham; 2020:323-344.
doi - Steven H, Swerdlow EC, Harris NL, Jaffe ES, Pileri SA. In: Stein H, Thiele J, Arber DA, Hasserjian RP, Le Beau MM, Orazi A, Siebert R, eds. WHO classification of tumours of haemaopoietic and lymphoid tissues. Lyon: WHO Press; 2017.
- Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364(24):2305-2315.
doi pubmed - Troussard X, Cornet E. Hairy cell leukemia 2018: Update on diagnosis, risk-stratification, and treatment. Am J Hematol. 2017;92(12):1382-1390.
doi pubmed - Chung SS, Kim E, Park JH, Chung YR, Lito P, Teruya-Feldstein J, Hu W, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med. 2014;6(238):238ra271.
doi - Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol. 1998;16(8):2780-2795.
doi pubmed - Campo E, Rule S. Mantle cell lymphoma: evolving management strategies. Blood. 2015;125(1):48-55.
doi pubmed - Furtado M, Rule S. Indolent mantle cell lymphoma. Haematologica. 2011;96(8):1086-1088.
doi pubmed - Ye H, Desai A, Zeng D, Nomie K, Romaguera J, Ahmed M, Wang ML. Smoldering mantle cell lymphoma. J Exp Clin Cancer Res. 2017;36(1):185.
doi pubmed - Martin P. A tale of two mantle cell lymphomas. Blood. 2018;132(4):347-348.
doi pubmed - Espinet B, Ferrer A, Bellosillo B, Nonell L, Salar A, Fernandez-Rodriguez C, Puigdecanet E, et al. Distinction between asymptomatic monoclonal B-cell lymphocytosis with cyclin D1 overexpression and mantle cell lymphoma: from molecular profiling to flow cytometry. Clin Cancer Res. 2014;20(4):1007-1019.
doi pubmed - Fernandez V, Salamero O, Espinet B, Sole F, Royo C, Navarro A, Camacho F, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res. 2010;70(4):1408-1418.
doi pubmed - Orchard J, Garand R, Davis Z, Babbage G, Sahota S, Matutes E, Catovsky D, et al. A subset of t(11;14) lymphoma with mantle cell features displays mutated IgVH genes and includes patients with good prognosis, nonnodal disease. Blood. 2003;101(12):4975-4981.
doi pubmed - Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. French-American-British (FAB) Cooperative Group. J Clin Pathol. 1989;42(6):567-584.
doi pubmed - Royo C, Navarro A, Clot G, Salaverria I, Gine E, Jares P, Colomer D, et al. Non-nodal type of mantle cell lymphoma is a specific biological and clinical subgroup of the disease. Leukemia. 2012;26(8):1895-1898.
doi pubmed - Haque M, Hameed N, Polski J. Composite peripheral T-Cell lymphoma and B-cell lymphoma with immunophenotype of hairy cell leukemia: report of an unusual case. Am J Clin Pathol. 2018;150:S93-S115.
doi - Gasljevic G, Kloboves-Prevodnik V, Gazic B, Vovk M. Coexistent hairy cell leukaemia and hepatosplenic T-cell lymphoma: a case report. Diagn Pathol. 2014;9:58.
doi pubmed - Paolini R, Poletti A, Ramazzina E, Menin C, Santacatterina M, Montagna M, Bonaldi L, et al. Co-existence of cutaneous T-cell lymphoma and B hairy cell leukemia. Am J Hematol. 2000;64(3):197-202.
doi - Liptrot S, D OB, Langabeer SE, Quinn F, Mackarel AJ, Elder P, Vandenberghe E, et al. An immunophenotypic and molecular diagnosis of composite hairy cell leukaemia and chronic lymphocytic leukaemia. Med Oncol. 2013;30(4):692.
doi pubmed - Rastogi P, Jeyaraman P, Sachdeva MU, Malhotra P, Ahluwalia J. Synchronous hairy cell leukemia and chronic lymphocytic leukemia: a case report with a brief review of literature. Blood Res. 2018;53(2):160-163.
doi pubmed - Obiorah IE, Francischetti IMB, Wang HW, Ahn IE, Wang W, Raffeld M, Kreitman RJ, et al. Concurrent chronic lymphocytic leukemia/small lymphocytic lymphoma and hairy cell leukemia: clinical, pathologic and molecular features. Leuk Lymphoma. 2020;61(13):3177-3187.
doi pubmed - Sychevskaia K, Kravchenko S, Misyurina A, Biderman B, Kovrigina A, Smirnova S, Nikulina E, et al. Molecular analysis of a composite B-cell tumor- hairy cell leukemia and mantle cell lymphoma combination. Hemasphere. 2019;3:S1.
doi - Gorodetskiy VR, Probatova NA, Kupryshina NA, Palshina SG, Obukhova TN, Sidorova YV, Ryzhikova NV, et al. Simultaneous presentation of leukemic non-nodal mantle cell lymphoma and gamma-delta T-large granular lymphocytic leukemia in a patient with rheumatoid arthritis. Cancer Manag Res. 2020;12:9449-9457.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.