

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 10, Number 4, August 2021, pages 202-205

Severe Vitamin B12 Deficiency Mimicking Microangiopathic Hemolytic Anemia
Ramzi Hassouneha, f, Steve Shenb, Olivia Leeb, Rachel A. Hartc, Logan P. Rhead, Patrick Faddena, e
aDepartment of Internal Medicine, Virginia Commonwealth University Health System, Richmond, VA 23298, USA
bVirginia Commonwealth University School of Medicine, Richmond, VA 23298, USA
cDepartment of Internal Medicine, East Carolina University/Vidant Medical Center, Greenville, NC 27834, USA
dDepartment of Hematology, Oncology, and Palliative Care, Virginia Commonwealth University Health System, Richmond, VA 23298, USA
eHunter Holmes McGuire Veterans Affairs Medical Center, Richmond, VA 23249, USA
fCorresponding Author: Ramzi Hassouneh, Department of Internal Medicine, Virginia Commonwealth University, 1200 East Broad Street, PO Box 980135, Richmond, VA 23298, USA
Manuscript submitted July 11, 2021, accepted July 31, 2021, published online August 30, 2021
Short title: Vitamin B12 Deficiency Mimicking MAHA
doi: https://doi.org/10.14740/jh889
| Abstract | ▴Top |
Most individuals with vitamin B12 deficiency present with anemia, fatigue, and neurologic disturbances such as paresthesia and loss of sensory function if chronic. However, in severe states, it may manifest as hemolytic anemia, thrombocytopenia, schistocytosis, elevated lactate dehydrogenase, and low reticulocyte production. This phenomenon is known as pseudo-thrombotic microangiopathy (TMA), and is most commonly due to pernicious anemia. The overlap in clinical presentation with primary TMA creates a challenge in the diagnosis and management of pseudo-TMA. Primary TMA, particularly thrombotic thrombocytopenic purpura, is emergently managed with plasma exchange and may require admission to an intensive care unit due to high risk of mortality. In contrast, pseudo-TMA does not respond to plasma exchange and instead is treated with vitamin B12 supplementation. Patients with this atypical presentation of B12 deficiency may receive unnecessary, costly, and potentially harmful therapy. We present the case of a patient with pseudo-TMA in the setting of pernicious anemia.
Keywords: Vitamin B12 deficiency; Microangiopathic hemolytic anemia; Pseudo-thrombotic microangiopathy
| Introduction | ▴Top |
Thrombotic microangiopathy (TMA) is a severe but rare condition characterized by microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and possible organ dysfunction. This process is associated with high mortality and is most commonly caused by disseminated intravascular coagulation (DIC) followed by thrombotic thrombocytopenia purpura (TTP) and hemolytic uremic syndrome (HUS) [1]. MAHA refers to anemia from Coombs negative red blood cell hemolysis with evidence of schistocytes on peripheral blood smear due to intravascular destruction. Interestingly, severe vitamin cobalamin (B12) deficiency may present similarly with approximately 2.5% of patients undergoing what is known as pseudo-TMA [2]. Pseudo-TMA is characterized by hemolytic anemia, thrombocytopenia, schistocytosis, elevated lactate dehydrogenase (LDH), and low reticulocyte production [3, 4].
Given the overlap in presentation, differentiating between primary TMA and pseudo-TMA remains a diagnostic challenge. It is important to consider severe B12 deficiency as a cause because the treatment and prognoses are vastly different. Primary TTP is emergently managed with plasma exchange and may require admission to an intensive care unit as it is associated with a high mortality if left untreated. On the other hand, pseudo-TMA due to severe B12 deficiency can simply be treated with vitamin supplementation [3, 4]. The potential for misdiagnosis can lead to complications from unnecessary and possibly harmful therapy and higher cost to the individual.
| Case Report | ▴Top |
A 73-year-old African American man with no known past medical history presented with 2 weeks of weakness, fatigue, and dyspnea on exertion. He endorsed loss of taste and increasing anorexia which subsequently led to noticeable weight loss. He denied a personal or family history of hematologic or coagulation disorders, autoimmune disease, or malignancy. He endorsed heavy alcohol consumption in his 20s and 30s, but denied recent alcohol, tobacco, or illicit drug use.
On admission, his vital signs were significant for a heart rate of 115 bpm. On physical exam, he was noted to have pale conjunctiva, a yellow soft palate, and a swollen tongue consistent with glossitis. Cardiovascular exam was significant for a loud mid-systolic ejection murmur best heard at the left sternal border. His skin exam was negative for petechiae or rashes. Sensation to light touch, temperature, and vibration as well as motor function was intact bilaterally with no focal deficits. Complete blood count (CBC) revealed hemoglobin of 5.3 g/dL, mean corpuscular volume (MCV) of 106.2 fL, and platelet count of 81,000/µL. The hepatic panel was significant for an unconjugated hyperbilirubinemia of 1.2 mg/dL (total bilirubin 1.5 mg/dL). Further laboratory workup was remarkable for severe vitamin B12 deficiency less than 60 pg/mL, elevated LDH greater than 4,000 U/L, haptoglobin less than 31 mg/dL, and negative direct antiglobulin test. Iron studies and folate level were within normal limits. Methylmalonic acid was elevated at 3,460 nmol/L. Additionally, reticulocyte production index (RPI) was low at 0.47, suggestive of a hypoproliferative bone marrow and not typical for hemolytic anemia which commonly presents with a state of hyperproliferation. A review of his peripheral blood smear showed hypersegmented neutrophils, nucleated red blood cells, schistocytes, tear drop cells, and anisopoikilocytosis (Fig. 1).
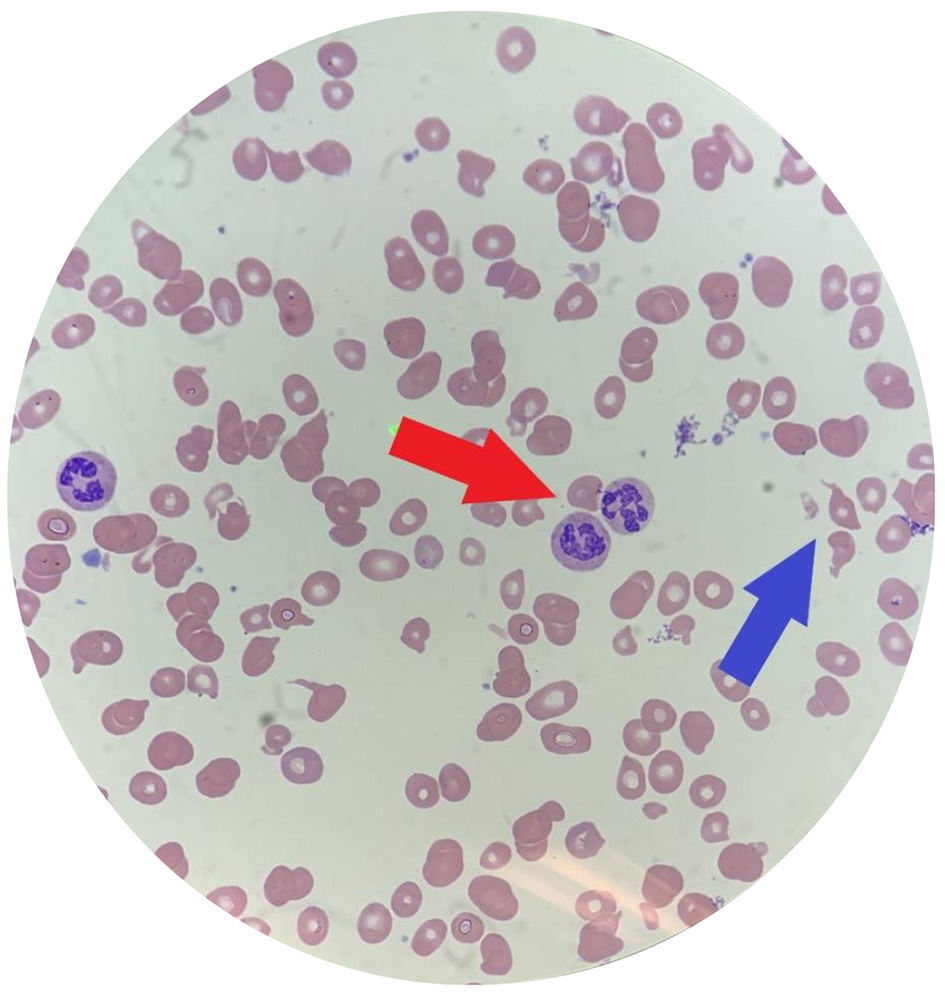 Click for large image | Figure 1. Peripheral blood smear of our patient. Note multiple hypersegmented neutrophils (red arrow) and schistocytes (blue arrow). Additionally, this smear is significant for marked anisocytosis, poikilocytosis, macrocytosis and hypochromia. |
The patient received a total of four units of packed red blood cells during his first 24 h of admission to achieve a goal hemoglobin above 7 g/dL while his workup was pending. On day 2 of admission, he was started on daily 1,000 µg intramuscular vitamin B12 once his severe vitamin B12 deficiency was confirmed. Over the next 3 days, he reported resolution of his symptoms. His hemoglobin continued to improve and remained relatively stable at 9 g/dL for the remainder of his admission. Notably no schistocytes were detected on blood smear by day 3 of admission compared to 1-5% schistocytes per high-power field on days 1 and 2. Due to improvement of his symptoms and hemoglobin, bone marrow biopsy was not performed. Prior to discharge, the patient underwent an esophagoduodenoscopy which showed no evidence of atrophic gastritis. He was tested for intrinsic factor auto-antibodies and was found to be positive approximately 1 week after discharge, consistent with pernicious anemia. After a 4-day hospital course, he was discharged with a prescription for 1,000 µg intramuscular vitamin B12 to be taken daily for 14 days. At 2-week follow-up, he showed improvement of his anemia with a hemoglobin of 10.1 g/dL (MCV 97.9 fL). At this outpatient visit, he was placed on monthly 1,000 µg of intramuscular vitamin B12. At 3-month follow-up, he had complete resolution of his anemia with a hemoglobin of 13.4 g/dL (MCV 82.3 fL). His laboratory data are summarized in Table 1.
 Click to view | Table 1. Summary of Laboratory Data During Hospitalization and Follow-Up |
| Discussion | ▴Top |
Vitamin B12 is a water-soluble vitamin that is abundant in common foods such as dairy products, meats, and fish. During digestion, B12 forms a complex with intrinsic factor produced by gastric parietal cells and is then absorbed by the terminal ileum. Vitamin B12 plays a critical role in DNA synthesis, the division and maturation of hematopoietic cells, and normal neurologic function [5]. It serves as a cofactor for three important enzymatic reactions: the conversion of 5-methyltetrahydrofolate to tetrahydrofolate, the conversion of methylmalonic acid to succinyl coenzyme A, and the conversion of homocysteine to methionine [5-7].
Vitamin B12 deficiency is common and occurs in 20% of people older than 60 years of age [5]. The most common causes of B12 deficiency include inadequate dietary intake in the pediatric population and pernicious anemia in adults [8]. Pernicious anemia is a hematologic manifestation of autoimmune gastritis characterized by autoantibody formation against intrinsic factor or gastric parietal cells [9]. This was seen in our patient with the presence of anti-intrinsic factor antibodies.
Multiple organ systems can be affected to varying degrees during vitamin B12 deficiency. The accumulation of methylmalonic acid contributes to progressive demyelination with neurologic manifestations such as loss of proprioception and vibratory sensation, peripheral neuropathy, and ataxia [5, 7]. DNA synthesis is altered due to inability to form tetrahydrofolate and this most commonly manifests as megaloblastic anemia with MCV greater than 100 fL, but can also cause bone marrow failure and result in thrombocytopenia, neutropenia, and even pancytopenia. Hyperhomocysteinemia has potential hemolytic effects through the generation of reactive oxygen species, lipid peroxidation of cell membranes, endothelial damage, platelet aggregation, and coagulation activation [7, 10]. This results in microangiopathy with fragmentation of erythrocytes and schistocytosis. This very rare finding is known as pseudo-TMA and is found in less than 2.5% of patients with severe vitamin B12 deficiency [11].
It is important that pseudo-TMA is differentiated from primary TMA. TTP, a type of primary TMA, is a life-threatening disorder that needs to be ruled out in patients presenting with hemolytic anemia, schistocytosis, and thrombocytopenia. TTP is caused by severely reduced activity of the von Willebrand factor-cleaving protease ADAMTS13 [12-15]. This leads to formation of platelet microthrombi with ensuing microvascular insult to the brain, kidney, or heart. TTP is a medical emergency and without treatment, mortality from TTP is approximately 80-90% [12]. Treatment for TTP involves prompt plasma exchange which may require admission to an intensive care unit, whereas pseudo-TMA is unresponsive to plasma exchange and is simply treated with B12 supplementation. A systematic review of 41 cases of B12 deficiency presenting with TMA found that 14 patients received unnecessary plasma exchange [16]. Two cases were associated with major complications which included anaphylactic transfusion reaction in one and hemothorax and cardiac arrest due to line placement in the other. This highlights the need to diagnose pseudo-TMA swiftly so that patients do not receive costly, ineffective therapy and be at risk for potential adverse events [15].
Given the rarity of pseudo-TMA, diagnosis can be a challenge. Although the laboratory findings of pseudo-TMA are similar to TTP, there are some notable differences. The reticulocyte production index is important in discriminating between pseudo-TMA and TTP [13-15]. A compensatory increase in erythropoiesis is typically observed in primary TMA syndromes as there is no shortage of substrates for DNA synthesis, unlike in nutritional deficiencies. This is reflected with an elevated reticulocyte production index (RPI) and in TTP the RPI is routinely greater than 3 [17]. However, in pseudo-TMA, compensatory increase in erythropoiesis is not observed. The lack of B12 causes impaired DNA synthesis and suppression to bone marrow production of hematopoietic cells. Therefore, the bone marrow is unable to appropriately increase reticulocyte production to replace lost red blood cells. The RPI in pseudo-TMA is typically less than 2 which indicates an inadequate response by the bone marrow [17]. This is consistent with our patient’s low RPI of 0.47.
High levels of LDH are observed in pseudo-TMA but not in TTP because LDH is a byproduct of nucleated cells. Vitamin B12 deficiency in pseudo-TMA causes dysfunctional DNA synthesis, arrest in cellular division, and subsequent intramedullary cell death [18]. LDH is then released in high quantities during the intramedullary hemolysis of immature nucleated erythrocyte precursors in the bone marrow. Contrastingly, hemolysis in TTP occurs in peripheral blood with intravascular destruction of mature non-nucleated erythrocytes. Studies show that LDH is routinely 2,500 IU/L or greater in pseudo-TMA and less than 2,500 IU/L in TTP [13]. In our case report, the patient’s LDH level exceeded 4,000 IU/L.
There is only modest unconjugated hyperbilirubinemia in pseudo-TMA compared to TTP [3]. Unconjugated bilirubin is a byproduct of heme degradation. The intramedullary destruction of erythrocyte precursors produces little hemoglobin and so there is only minor elevation in bilirubin levels [14]. Our patient’s unconjugated bilirubin level upon presentation was 1.5 mg/dL. Whereas in TTP, the intravascular hemolysis of mature erythrocytes, which contain a higher concentration of hemoglobin compared to precursor cells, in peripheral blood results in significantly higher levels of unconjugated bilirubin [14].
Learning points
Severe vitamin B12 deficiency as demonstrated in our patient, can present with a rare disorder known as pseudo-TMA that mimics primary TMA syndromes with similar features of hemolytic anemia, thrombocytopenia, and schistocytes. This presentation may be unrecognized and may place patients with pseudo-TMA due to severe B12 deficiency at risk for unnecessary plasma exchange. Understanding the important distinguishing features of pseudo-TMA including low RPI and high LDH relative to unconjugated bilirubin levels can prompt clinicians to begin rapid B12 supplementation for improvement in clinical status.
Acknowledgments
None to declare.
Financial Disclosure
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
None to declare.
Informed Consent
Informed consent has been obtained from the patient for this manuscript.
Author Contributions
Ramzi Hassouneh: drafting manuscript, critical revisions, and final approval. Steve Shen: drafting manuscript, critical revisions, and final approval. Olivia Lee: drafting manuscript, critical revisions, and final approval. Rachel Hart: drafting manuscript, critical revisions, and final approval. Logan Rhea: pathology interpretation, critical revisions, and final approval. Patrick Fadden: critical revisions and final approval.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Scully M, Cataland S, Coppo P, de la Rubia J, Friedman KD, Kremer Hovinga J, Lammle B, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312-322.
doi pubmed - Antony AC. Hematology, basic principles and practice. Philadelphia, PA, Elsevier; 2005.
- Noel N, Maigne G, Tertian G, Anguel N, Monnet X, Michot JM, Goujard C, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM. 2013;106(11):1017-1022.
doi pubmed - Tun AM, Myint ZW, Hernandez CR, Guevara E, Thein KZ, Oo TH. Vitamin B12 deficiency related pseudo-thrombotic microangiopathy might be misdiagnosed and treated with plasma product therapy: review of the literature and analysis of the reported cases. Blood. 2017;130:5576.
- Langan RC, Goodbred AJ. Vitamin B12 deficiency: recognition and management. Am Fam Physician. 2017;96(6):384-389.
- Shipton MJ, Thachil J. Vitamin B12 deficiency - A 21st century perspective. Clin Med (Lond). 2015;15(2):145-150.
doi pubmed - Jarquin Campos A, Risch L, Nydegger U, Wiesner J, Vazquez Van Dyck M, Renz H, Stanga Z, et al. Diagnostic accuracy of holotranscobalamin, Vitamin B12, methylmalonic acid, and homocysteine in detecting B12 deficiency in a large, mixed patient population. Dis Markers. 2020;2020:7468506.
doi pubmed - Stabler SP, Allen RH. Vitamin B12 deficiency as a worldwide problem. Annu Rev Nutr. 2004;24:299-326.
doi pubmed - Katiyar V, Qian E, Vohra I, Sleiman J, Rubinstein P. Pernicious anemia presenting with pseudo thrombotic microangiopathy and falsely elevated B 12 levels. J Hematol. 2019;8(3):129-131.
doi pubmed - Ventura P, Panini R, Tremosini S, Salvioli G. A role for homocysteine increase in haemolysis of megaloblastic anaemias due to vitamin B(12) and folate deficiency: results from an in vitro experience. Biochim Biophys Acta. 2004;1739(1):33-42.
doi pubmed - Andres E, Affenberger S, Zimmer J, Vinzio S, Grosu D, Pistol G, Maloisel F, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006;28(1):50-56.
doi pubmed - Chaturvedi S, Carcioppolo D, Zhang L, McCrae KR. Management and outcomes for patients with TTP: analysis of 100 cases at a single institution. Am J Hematol. 2013;88(7):560-565.
doi pubmed - Walter K, Vaughn J, Martin D. Therapeutic dilemma in the management of a patient with the clinical picture of TTP and severe B12 deficiency. BMC Hematol. 2015;15:16.
doi pubmed - Garderet L, Maury E, Lagrange M, Najman A, Offenstadt G, Guidet B. Schizocytosis in pernicious anemia mimicking thrombotic thrombocytopenic purpura. Am J Med. 2003;114(5):423-425.
doi - Adamski J. Thrombotic microangiopathy and indications for therapeutic plasma exchange. Hematology Am Soc Hematol Educ Program. 2014;2014(1):444-449.
doi pubmed - Tran PN, Tran MH. Cobalamin deficiency presenting with thrombotic microangiopathy (TMA) features: A systematic review. Transfus Apher Sci. 2018;57(1):102-106.
doi pubmed - Trubin PA, Edward JA, Hand M. Pseudo-thrombotic thrombocytopenic purpura due to severe vitamin B12 deficiency. J La State Med Soc. 2016;168(6):196-200.
- Rao S, Colon Hidalgo D, Doria Medina Sanchez JA, Navarrete D, Berg S. Et Tu, B12? Cobalamin deficiency masquerading as pseudo-thrombotic microangiopathy. Cureus. 2020;12(7):e9097.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.