

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Original Article
Volume 10, Number 3, June 2021, pages 106-113

Essential Thrombocythemia in Children: A Retrospective Study
Muhammed Ameena, Khawar Siddiquia, Saadiya Khana, Mahasen Saleha, Abdullah Al-Jefria, Abdulrahman Al-Musaa, b
aDepartment of Pediatric Hematology/Oncology, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
bCorresponding Author: Abdulrahman Al-Musa, Department of Pediatric Hematology/Oncology, King Faisal Specialist Hospital and Research Center, Riyadh 11211, Saudi Arabia
Manuscript submitted March 28, 2021, accepted April 17, 2021, published online June 16, 2021
Short title: Essential Thrombocythemia in Pediatric Patients
doi: https://doi.org/10.14740/jh822
| Abstract | ▴Top |
Background: Essential thrombocythemia (ET) is one of the “classic” Philadelphia chromosome negative (Ph-) myeloproliferative neoplasms characterized by sustained thrombocytosis, increased megakaryopoiesis and high risk of vascular complications. ET is very rare in childhood. The annual incidence is approximately 1 per 10,000,000 in children less than 14 years, and about 60 times lower than adults. The genetic landscape and clonal features in childhood ET has not been well defined. There is no evidence-based guidance on the diagnosis of childhood ET.
Methods: Medical records of 28 pediatric patients (age ≤ 14 years at diagnosis) with ET were reviewed and evaluated to characterize the different mutation profiles and to evaluate the treatment modalities used and the potential long-term outcome.
Results: More than half of the patients were found to have positive history of parental consanguinity (57.1%) whereas positive family history was documented for more than a quarter of our patients (28.6%). Janus kinase 2 gene (JAK2) V617F mutation was positive in two of 26 patients (7.7%). Myeloproliferative leukemia virus oncogene (MPL) exon 10 and calreticulin (CALR) mutations were tested in eight patients, which were negative for all of them. Treatment included low-dose aspirin (LDA) in seven patients (50%), combination of LDA with hydroxyurea in three patients (21.4%), hydroxyurea in two patients (14.3%), combination of platelets apheresis with LDA and anagrelide in one patient each (7.1%). During the treatment, two patients experienced stroke (7.1%), one patient developed Budd-Chiari syndrome (3.6%) and one patient developed azoospermia (3.6%).
Conclusions: The incidence of ET in children is extremely low in Saudi Arabia. Most of the children with ET were asymptomatic, and thrombocytosis was often discovered incidentally. JAK2 V617F mutation has no known impact on the prognosis or on the outcome of the disease in the pediatric age group that is in contrast to the adult ET. Children less than 1 year are at high risk for complications particularly during acute precipitating infectious episode. The potential complications and clinical course of pediatric ET are unpredictable.
Keywords: Essential thrombocythemia; Pediatric ET; JAK2 mutation
| Introduction | ▴Top |
Essential thrombocythemia (ET) is one of the subtypes of breakpoint cluster region protein/Abelson murine leukemia viral oncogene homolog 1 (BCR/ABL1)-negative myeloproliferative neoplasms (MPNs). It is a clonal hematopoietic stem cell disorder that is characterized by isolated thrombocytosis and is associated with complications such as thrombosis, hemorrhage, and progression to myelofibrosis or acute myeloid leukemia. ET is a disorder occurring predominantly in middle-age adults. The disorder is very rare in childhood and adolescence [1-3]. The annual incidence of ET is approximately 1 per 10,000,000 in children less than 14 years and about 60 times lower than that of adults [4-6].
The past decade and a half has enabled us to understand ET better due to the developments made in the field of molecular pathogenesis. Discovery of Janus kinase 2 gene (JAK2) V617F in 2005, followed by myeloproliferative leukemia virus oncogene (MPL) mutations in 2006 and calreticulin (CALR) mutations in 2013 has contributed to a more subtle understanding of disease pathology, diversity of clinical presentation and diagnostic possibilities [7-11]. The most frequent driver mutation seen is JAK2 V617F, which is found in about 99% of patients with polycythemia vera (PV), 55% ET, and 65% in primary myelofibrosis (PMF) [12]. The genetic landscape and clonal features in childhood ET has not been well defined.
For a diagnosis of ET to be consistent with the World Health Organization (WHO) criteria it has to have a platelet count of ≥ 450 × 109/L, presence of one of the three above mentioned driver mutations or in their absence the exclusion of other causes of thrombocytosis (reactive and clonal), and bone marrow morphologic assessment, especially for distinguishing ET from prefibrotic PMF and “masked” PV [13, 14]. In addition to clonal thrombocytosis, some patients with ET can have clinical signs of mild splenomegaly, leukocytosis, microvascular symptoms, thrombotic and bleeding complications while running the risk of leukemic transformation or fibrotic progression as time goes on.
Herein we present our retrospective analysis of 28 patients treated at our institution. Our aim was to evaluate and characterize the different mutation profiles associated with ET in our indigenous population and assess the treatment modalities used along with their potential long-term outcome.
| Materials and Methods | ▴Top |
A retrospective case study was conducted at the King Faisal Specialist Hospital and Research Center (KFSHRC), Saudi Arabia. Medical records of 28 pediatric patients (age ≤ 14 years at diagnosis) with ET from January 1987 to December 2017 were reviewed and evaluated. Data on demographics, diagnosis, hematological parameters and outcome were obtained from clinical databases and through medical charts review. After quality assurance checks, the dataset was then transferred to IBM-SPSS for Windows Version 20.0 for final analysis. Furthermore, a literature review of similar studies was conducted, and a comparison was drawn between the findings of our study and the previous literature.
Statistical considerations
All continuous data are presented as median with minimum and maximum points, while discrete data are provided as number (%). Independent-samples Mann-Whitney U test was utilized to test for significance of difference between genders for continuous data.
Ethical consideration
This study was submitted to the Institutional Review Board of KFSHRC, Riyadh, Saudi Arabia, and was approved by the Research Advisory Committee through established procedures with approval number 2181192.
This study was conducted following international guidelines and policies on conducting research on human subjects. Data of interest collected from the patients’ medical records were secured and governed by the institutional policies on patient confidentiality and privacy. All patient identifiers were masked at the time of data collection.
| Results | ▴Top |
This study showed a female to male ratio of 2.1:1, a female preponderance was observed. The median age at diagnosis was 8.8 years (range, at birth - 13.8 years). More than half of the patients were found to have positive history of parental consanguinity (n = 16, 57.1%). The patient’s characteristics and primary disease-related parameters are presented in Table 1.
 Click to view | Table 1. Demographics and Clinical Characteristics (N = 28) |
With a median follow-up time of 71.6 months, all our patients were alive at the last follow-up. Patient’s hematological parameters at presentation are provided in Table 2. The median platelet count at presentation was found to be 846.5 × 109/L with a range of 529 × 109/L to 2,003 × 109/L (Fig. 1). We did not observe statistically significant difference between the genders for all recorded hematological values, neither any measure of correlation was found to be statistically significant except for hemoglobin (Hb) and red blood cell (RBC) (Spearman’s rho = 0.755, P < 0.001, Fig. 2). Peripheral blood film confirmed thrombocytosis in all patients. Their workup included serum ferritin level, iron profile, prothrombin time (PT), activated partial thrombin time (PTT), von Willebrand level and function. PT and PTT were prolonged in 12 (52.2%) and five (21.7%) patients, respectively. Platelet function assay (PFA-100) was normal in all three patients, for whom it was performed. Bone marrow findings confirmed a hyperplastic megakaryocytic marrow, large and mature morphology with hyperlobated nuclei and abundant cytoplasm without myelofibrosis in all patients. Standard cytogenetic analysis for BCR/ABL rearrangement showed no detectable abnormalities. JAK2 V617F mutation was positive in two of 26 (7.7%) patients (Table 3). Clinical and laboratory features of these patients are provided in Table 4. Median platelet counts were significantly lower in JAK2 positive cases. Other than JAK2 V617F mutation, we checked for MPL exon 10 and CALR mutations in eight patients that was negative for all of them. Evaluation for von Willebrand (antigen and activity) was performed in all patients, and the level was normal in all patients (Table 2).
Click to view | Table 2. Hematological Profile at Presentation |
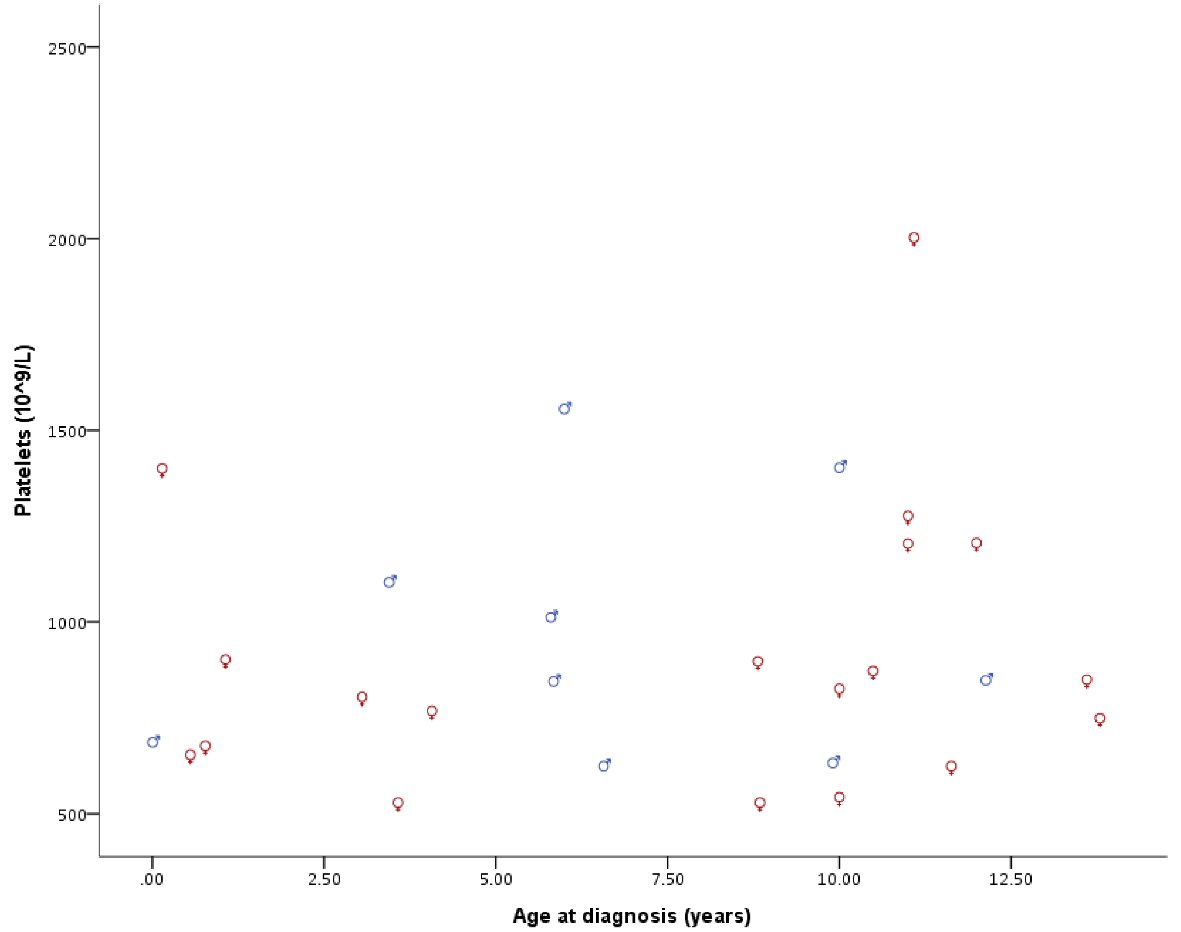 Click for large image | Figure 1. Platelet count at diagnosis by gender and age. |
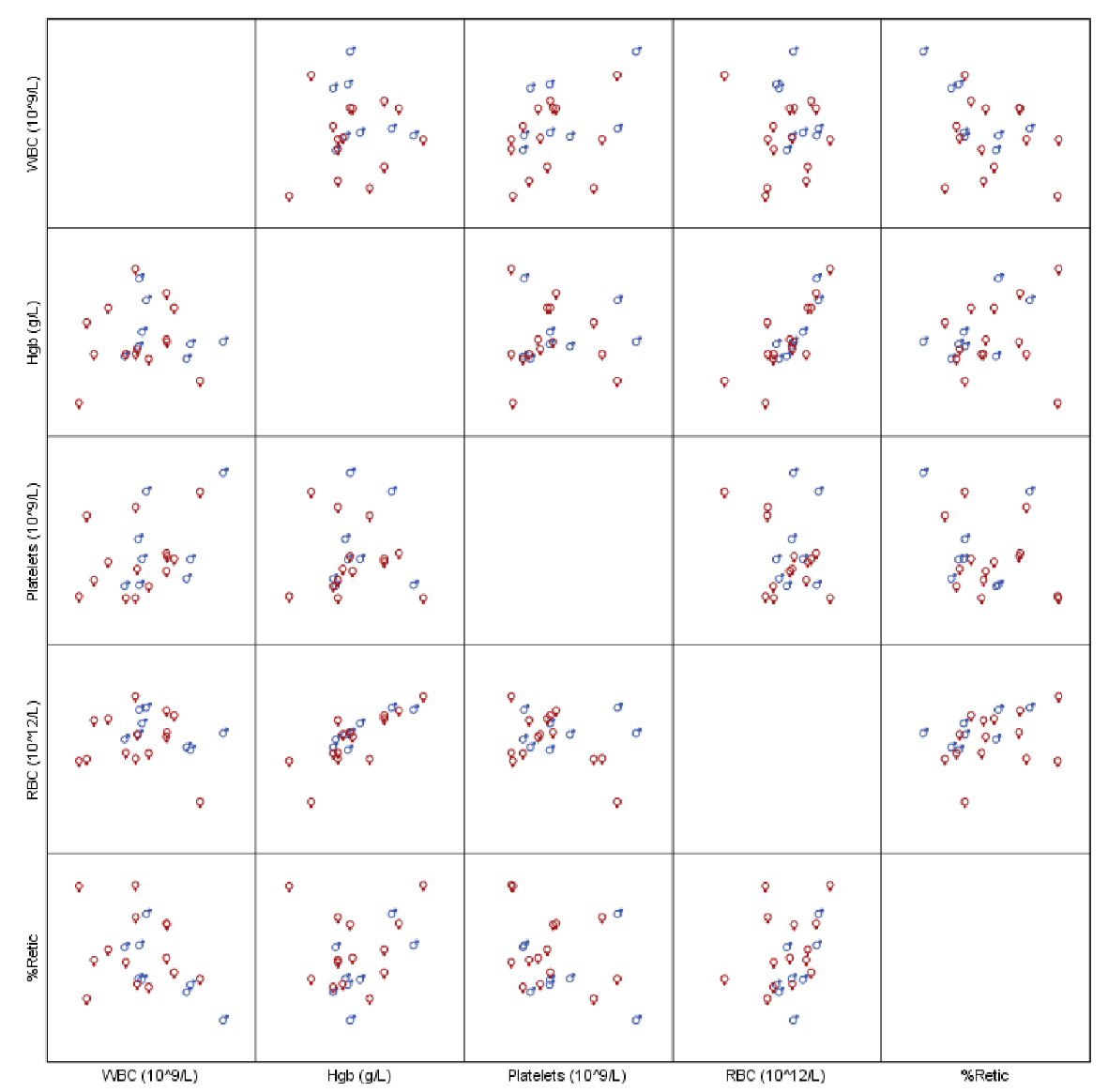 Click for large image | Figure 2. Scatterplot of hematological parameters of interest. |
Click to view | Table 3. Patient’s Profile at Presentation, Treatment Regimen and Follow-Up |
Click to view | Table 4. Clinical and Laboratory Featuresa by JAK2 Mutation Positivity (N = 26) |
With regards to treatment, half of the patients (n = 14) were offered treatment while the other half was kept under observation. Treatment included low-dose aspirin (LDA) in seven patients (50.0%), combination of LDA with hydroxyurea in three patients (21.4%), hydroxyurea alone in two patients (14.3%), combination of platelets apheresis with LDA and anagrelide in one patient each (7.1%). During the treatment and follow-up period, two of our patients experienced stroke (7.1%), one patient developed Budd-Chiari syndrome (3.6%) and one patient developed azoospermia (3.6%). The two patients who had stroke were on LDA, the patient who developed Budd-Chiari syndrome was on combination of platelets apheresis with LDA, while the patient who developed azoospermia was receiving hydroxyurea.
| Discussion | ▴Top |
The therapeutic goal in general for ET patients is to avoid the occurrence of major vascular events while minimizing the side effects induced by medication. Consensus regarding the management of adult ET follows a risk-adapted strategy. Currently, there is insufficient evidence to guide the management of childhood ET and usually a conservative approach is adopted. At our institution we treated our pediatric ET patients with LDA, if microcirculatory symptoms were present or the platelet counts were more than 1 million. Hydroxyurea or anagrelide was added, if the existing symptoms or the increased platelet counts did not improve. Otherwise, all other patients were managed conservatively with regular follow-up visits and complete blood count (CBC) every 3 - 6 months.
We studied a total of 28 patients diagnosed with ET. In our cohort of pediatric patients, the diagnosis of ET was made according to the 2016 WHO criteria in the absence of other known causes of reactive (secondary) thrombocytosis. Most of our patients (n = 20, 71.4%) were asymptomatic with high platelet counts with just a few of them (n = 8, 28.6%) experiencing symptoms. Our results on thrombocytosis in the absence of symptoms are comparable to others. Randi et al reported that majority (60%) of their patients were asymptomatic as well with other studies showing similar results [13]. Our results are also in line with published literature that has shown a low incidence of JAK2 V617F mutation in childhood ET [14-16].
During the follow-up period, four patients developed complications. Two patients experienced stroke, one was an infant who presented with viral infection with diarrhea at 3 months of age, while the second patient did so in adulthood following the diagnosis of ET. One of our patients developed Budd-Chiari syndrome around 15 years after diagnosis of ET. The last patient developed azoospermia that was probably secondary to the use of hydroxyurea. In our experience none of our patients had major complications with regards to leukemic or myelofibrotic transformation, which is in accordance with outcomes seen in the younger population [14, 17, 18]. High platelet count can lead to qualitative deficiency of von Willebrand factor (vWF) [19]. Acquired vWF disease can lead to bleeding in patients with ET. Bleeding was not a complication seen in any of our patients.
A limitation of our study is that it is a single institution data with retrospective analysis. Given the rarity of classical Philadelphia chromosome-negative myeloproliferative neoplasms (Ph- MPNs) disorders in children we believe that there should be both national and international collaborative efforts to study clinical and biologic parameters prospectively in these children. Knowing that majority of our studies patients did not carry the driver mutations we recommend a conservative approach for our indigenous population if there are no clinical concerns for pharmacologic intervention.
Conclusions
In conclusion, our experience shows that most of our children with ET were asymptomatic, and thrombocytosis was often discovered incidentally. In our practice pediatric patients are considered prone to complication particularly during acute infectious episodes that might place them at risk for dehydration and subsequent thrombosis. The potential complications and clinical course of pediatric ET can still be unpredictable.
Acknowledgments
None to declare.
Financial Disclosure
This work was not funded by any funding agency.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
No informed consents were obtained since this was a retrospective review; all data items collected were already documented in medical charts as part of the patient care and disease management documentation.
Author Contributions
Muhammed Ameen conducted the study, data collection through chart reviews, data entry, review of the results and manuscript preparation. Saadiya Khan: review of results and manuscript preparation and final approval of the manuscript. Khawar Siddiqui: study design, data management, processing and cleaning, primary data analysis, result presentation and reporting, and preparation of the manuscript. Mahasen Saleh: review of results, manuscript preparation and final approval of the manuscript. Abdullah Al-Jefri: review of results, manuscript preparation and final approval of the manuscript. Abdulrahman Al-Musa: conceptualization, study design, overall supervision of the project, review of the results, manuscript preparation and review as corresponding author with final approval of the manuscript, and as principal investigator of the study.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
Abbreviations
JAK2: Janus kinase 2 gene; CALR: calreticulin; MPL: myeloproliferative leukemia virus oncogene; BCR/ABL: breakpoint cluster region protein/Abelson murine leukemia viral oncogene; PT: prothrombin time; PTT: activated partial thrombin time; PFA-100: platelet function assay
| References | ▴Top |
- Cervantes F. Management of essential thrombocythemia. Hematology Am Soc Hematol Educ Program. 2011;2011:215-221.
doi pubmed - Hasle H. Incidence of essential thrombocythaemia in children. Br J Haematol. 2000;110(3):751.
doi pubmed - Barbui T. How to manage children and young adults with myeloproliferative neoplasms. Leukemia. 2012;26(7):1452-1457.
doi pubmed - Teofili L, Giona F, Martini M, Cenci T, Guidi F, Torti L, Palumbo G, et al. Markers of myeloproliferative diseases in childhood polycythemia vera and essential thrombocythemia. J Clin Oncol. 2007;25(9):1048-1053.
doi pubmed - Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054-1061.
doi - Ismael O, Shimada A, Hama A, Sakaguchi H, Doisaki S, Muramatsu H, Yoshida N, et al. Mutations profile of polycythemia vera and essential thrombocythemia among Japanese children. Pediatr Blood Cancer. 2012;59(3):530-535.
doi pubmed - James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148.
doi pubmed - Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472-3476.
doi pubmed - Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270.
doi pubmed - Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390.
doi pubmed - Tefferi A, Pardanani A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol. 2015;1(1):97-105.
doi pubmed - Barbui T, Thiele J, Gisslinger H, Finazzi G, Vannucchi AM, Tefferi A. The 2016 revision of WHO classification of myeloproliferative neoplasms: Clinical and molecular advances. Blood Rev. 2016;30(6):453-459.
doi pubmed - Kvasnicka HM, Orazi A, Thiele J, Barosi G, Bueso-Ramos CE, Vannucchi AM, Hasserjian RP, et al. European LeukemiaNet study on the reproducibility of bone marrow features in masked polycythemia vera and differentiation from essential thrombocythemia. Am J Hematol. 2017;92(10):1062-1067.
doi pubmed - Randi ML, Putti MC, Scapin M, Pacquola E, Tucci F, Micalizzi C, Zanesco L, et al. Pediatric patients with essential thrombocythemia are mostly polyclonal and V617FJAK2 negative. Blood. 2006;108(10):3600-3602.
doi pubmed - Giona F, Teofili L, Moleti ML, Martini M, Palumbo G, Amendola A, Mazzucconi MG, et al. Thrombocythemia and polycythemia in patients younger than 20 years at diagnosis: clinical and biologic features, treatment, and long-term outcome. Blood. 2012;119(10):2219-2227.
doi pubmed - Nakatani T, Imamura T, Ishida H, Wakaizumi K, Yamamoto T, Otabe O, Ishigami T, et al. Frequency and clinical features of the JAK2 V617F mutation in pediatric patients with sporadic essential thrombocythemia. Pediatr Blood Cancer. 2008;51(6):802-805.
doi pubmed - Alvarez-Larran A, Cervantes F, Bellosillo B, Giralt M, Julia A, Hernandez-Boluda JC, Bosch A, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia. 2007;21(6):1218-1223.
doi pubmed - Boddu P, Masarova L, Verstovsek S, Strati P, Kantarjian H, Cortes J, Estrov Z, et al. Patient characteristics and outcomes in adolescents and young adults with classical Philadelphia chromosome-negative myeloproliferative neoplasms. Ann Hematol. 2018;97(1):109-121.
doi pubmed - Awada H, Voso MT, Guglielmelli P, Gurnari C. Essential thrombocythemia and acquired von Willebrand Syndrome: the shadowlands between thrombosis and bleeding. Cancers (Basel). 2020;12(7):1746. Published 2020 Jun 30.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.