

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 10, Number 1, February 2021, pages 25-29
Histologic Transformation in an Untreated Waldenstrom’s Macroglobulinemia After 14 Years: Case Report and Review of the Literature
Elizabeth B. Elimimiana, Nadeem Bilania, Maria J. Diacovoa, Skirmante Sirvaitisb, Chieh Lin Fua, c
aDepartment of Hematology-Oncology, Maroone Cancer Center, Cleveland Clinic, Florida 2950 Cleveland Clinic Blvd, Weston, FL 33331, USA
bMercy Health, Internal Medicine Residency Program, Rockford, IL 61114, USA
cCorresponding Author: Chieh Lin Fu, Department of Hematology Oncology, Maroone Cancer Center, Cleveland Clinic Florida, 2950 Cleveland Clinic Blvd, Weston, FL 33331, USA
Manuscript submitted October 1, 2020, accepted November 14, 2020, published online February 6, 2021
Short title: HT in Untreated WM After 14 Years
doi: https://doi.org/10.14740/jh767
| Abstract | ▴Top |
Waldenstrom’s macroglobulinemia (WM) is an indolent B-cell non-Hodgkin lymphoma characterized by lymphoplasmacytic histology in the bone marrow with monoclonal IgM. Median survival can be in excess of 10 years. The 5-year cumulative incidence of death is low at about 10%. One-third of all-cause specific mortality is due to the lymphoma for which histologic transformation (HT) is rare. Here we present a case of a 60-year-old man with longstanding untreated WM, presenting with minimally symptomatic transformation to diffuse large B-cell lymphoma (DLBCL), with an accompanying review of the literature. Transformed WM, diagnosed greater than 5 years, has a reported survival period of 8 - 9 months. This case highlights that after a decade of continued stability in WM, not requiring treatment, an acute change in laboratory data with minimally progressive IgM levels, in the absence of B symptoms and clinical findings, may be the harbinger of transformation and at the time of diagnosis can have a rapidly deteriorating clinical course. In this case, the tripling of the lactate dehydrogenase (LDH) as the primary drastic change demonstrates the importance of the rapid increase in LDH as a singly reliable marker for HT. Late transformation has been borne out as a negative variable as the generally indolent course of WM is curtailed with the poor outcome in HT. Although MYD88 wildtype is a possible predictive factor for transformation, it is unclear if late transformation is clonally or non-clonally related and further molecular investigation is needed.
Keywords: Waldenstrom’s macroglobulinemia; Histologic transformation; Diffuse large B-cell lymphoma; Non-Hodgkin lymphoma
| Introduction | ▴Top |
Waldenstrom’s macroglobulinemia (WM) is an indolent B-cell non-Hodgkin lymphoma (NHL) representing 1-2% of all NHLs characterized by lymphoplasmacytic histology in the bone marrow, with monoclonal IgM [1, 2]. WM was first described in 1944 as having myeloma-like features, with proliferation of B cells causing increased serum IgM that can manifest as hyperviscosity syndrome. These include bleeding or thrombosis that can be associated with cryoglobulinemia, autoimmune hemolytic anemia and neuropathy without corresponding osteolysis, associated with lymphadenopathy and splenomegaly [2-4]. The MYD88 L265P mutation is detected in 90% of WM [1]. Median age at diagnosis is 70; median survival can be in excess of 10 years; the 5-year cumulative incidence of death is low at about 10% [5]. One-third of all-cause specific mortality is due to the lymphoma [6], for which histologic transformation (HT) is rare.
Here we present a case of a 60-year-old man with longstanding untreated WM, presenting with minimally symptomatic transformation to diffuse large B-cell lymphoma (DLBCL), with an accompanying review of the literature.
| Case Report | ▴Top |
The patient was a 60-year-old man who had an IgM kappa monoclonal gammopathy with normal complete blood cell count (CBC) and IgM levels stable, at approximately 400 mg/dL (40 - 230 mg/dL) for 14 years, monitored by observation [7]. However, he developed progression of WM manifested as pancytopenia. CBC revealed white blood cell count (WBC) of 3.7 × 103/µL (3.8 - 10.8 × 103/µL), hemoglobin of 9.4 g/dL (13.2 - 17.1 g/dL) and platelets of 46 × 103/µL(140 - 400 × 103/µL). CBC was normal 5 months prior. The IgM level was 666 mg/dL; lactate dehydrogenase (LDH) level was normal at 150 U/L (135 - 225 U/L) (Fig. 1). He denied constitutional “B symptoms”, including fever > 100.4 °F, drenching night sweats and weight loss. Repeat bone marrow aspiration and biopsy reported sheets of atypical small mature lymphoid cells, positive for PAX-5 and negative for cyclin-D1, with rare CD138-positive plasma cells (Fig. 2). Cytogenetics was normal. MYD88 mutation was detected. Flow cytometry revealed B cells with an abnormal immunophenotype that expressed CD11C (subset), CD19, CD20, CD23 (subset), CD45 and monotypic kappa surface immunoglobulin light chains. A computed tomography (CT) scan showed no evidence of splenomegaly, with inguinal nodes up to 14 mm. He received four doses of weekly rituximab (375 mg/m2) uneventfully. Four weeks after completion of rituximab, the pancytopenia improved (WBC 3.3 × 103/µL, hemoglobin 10.8 g/dL and platelets 333 × 103/µL). IgM levels declined to 568 mg/dL. Eight weeks later, repeat rituximab was given with an infusion-related reaction, which precluded completion. Ibrutinib was planned; however, the patient had deferred. Labs remained stable (WBC 3.9 × 103/µL, hemoglobin 11.0 g/dL and platelets 305 × 103/µL). IgM level was 461 mg/dL.
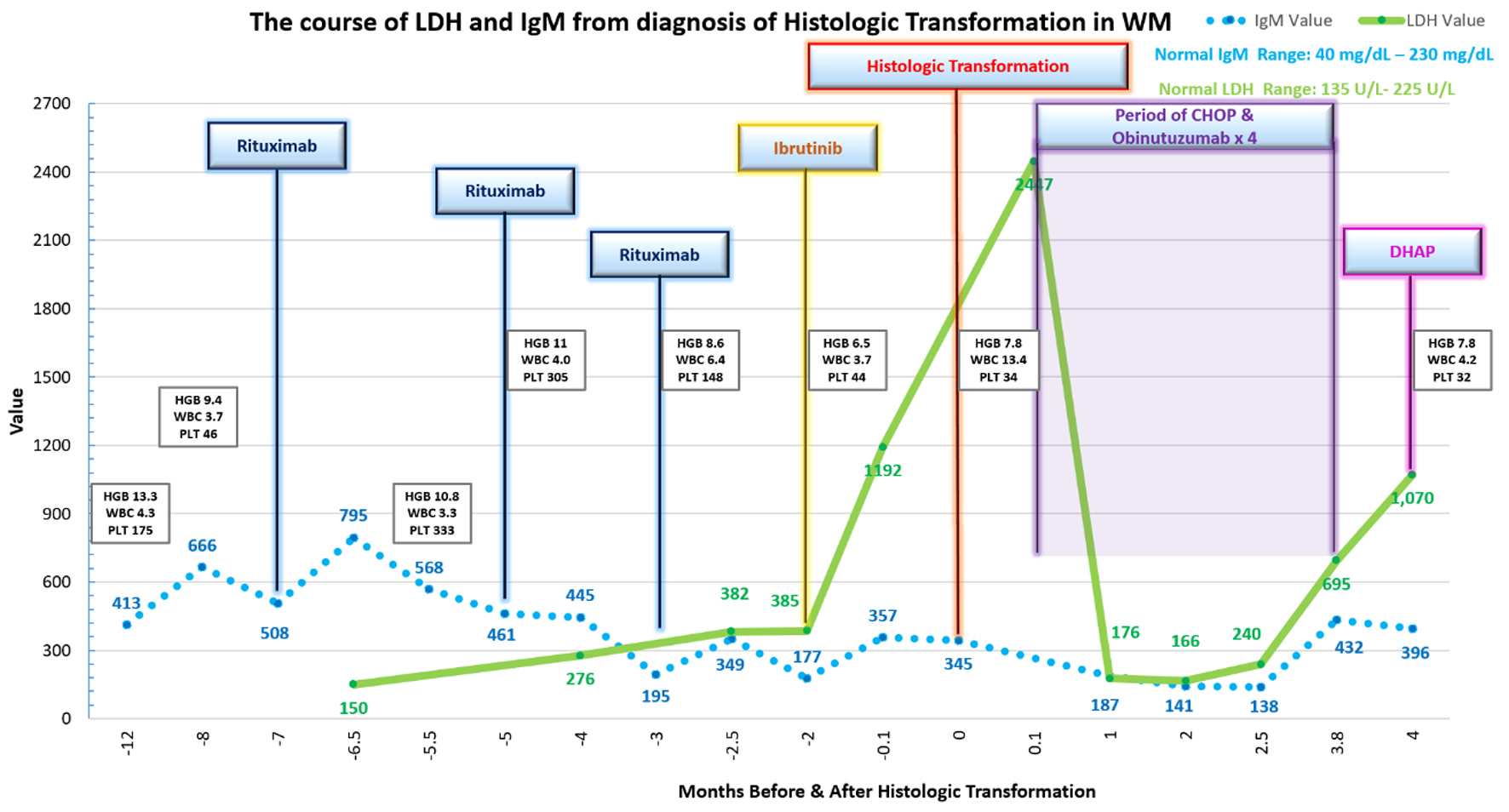 Click for large image | Figure 1. Diagram of histologic transformation course. WM: Waldenstrom’s macroglobulinemia; LDH: lactate dehydrogenase. |
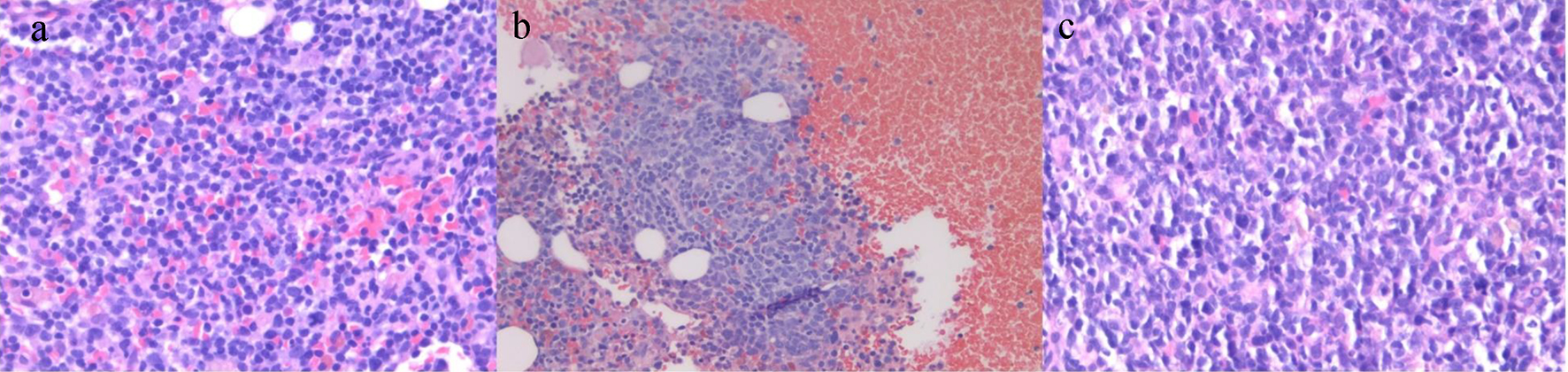 Click for large image | Figure 2. Pathology slides. (a) Bone marrow biopsy showing sheets of small atypical lymphoid cells and occasional plasma cells diffusely involving the bone marrow, consistent with Waldenstrom’s macroglobulinemia (H&E, × 40). (b) Bone marrow biopsy showing clusters of large malignant lymphoid cells in a background of bone marrow elements post-histologic transformation (H&E, × 20). (c) Lymph node biopsy showing large malignant lymphoid cells with centroblastic morphology. Histologic transformation to large cell lymphoma (H&E, × 40). H&E: hematoxylin and eosin. |
Rituximab treatment was attempted once more 8 weeks later resulting in serum sickness, which required hospital admission. At that time, labs revealed WBC of 6.4 × 103/µL, hemoglobin of 8.6 g/dL, platelets of 148 × 103/µL and IgM of 195 mg/dL. The peak interim LDH was 276 U/L.
Ibrutinib was initiated for progressive pancytopenia 4 weeks later (WBC 3.7 × 103/µL, hemoglobin 5.7 g/dL, platelets 44 × 103/µL and IgM 205 mg/dL). While on ibrutinib for 6 weeks, LDH levels tripled from 385 to 1,192 U/L. The patient still had no significant B symptoms at this time. Follow-up CT scans, in the interim 6 weeks, identified a right inguinal lymph node that increased from 2.4 to 3.8 cm. Spleen size increased from 11.4 to 14.2 cm. A positron emission tomography (PET) scan could not be done. Needle core biopsy of the inguinal lymph node revealed large atypical B cells expressing CD20, PAX-5, Bcl-2, MUM1 and high Ki-67 labeling index 80% diagnostic of HT to DLBCL. Immunohistochemical stains were negative for CD10, Bcl-6 and CD5. MYC positivity was present in approximately 30% of cells. Repeat bone marrow evaluation now revealed large cell lymphomatous involvement with complex cytogenetics (47,XY, add(3)(p13), add(6)(q27), add(8)(p21), del(9)(q13q22), +19[3]/48, idem, +mar[4]/47, idem, -add(6q), add(6) (p23)[3]/46,XY[10]).
He was planned to receive induction chemotherapy with hematopoietic stem cell transplantation (HSCT) and was offered a clinical trial of chimeric antigen receptor (CAR) T-cell therapy at the time. The decision was made for induction chemotherapy with consideration for HSCT if cytoreduction was achieved and CAR T-cell therapy if refractoriness. He was initiated on combination of CHOP and obinutuzumab, for four cycles requiring dose modifications for pancytopenia. He had progression on induction chemotherapy with an accelerated decline in performance status for which he was not eligible for CAR T-cell therapy. He had respiratory and constitutional symptoms with progressive pancytopenia and rising LDH consistent with progression of transformed lymphoma for which modified DHAP chemotherapy was initiated. During the salvage chemotherapy, he passed away with respiratory decline 4 months after diagnosis of HT.
| Discussion | ▴Top |
HT of WM to an aggressive lymphoma was first described in the 1960s [8, 9] with DLBCL being the most common. Rare transformation to aggressive immunoblastic lymphoma, peripheral T-cell lymphoma and a plasma-type neoplasm have also been reported [10, 11], as well as transformation to Hodgkin-type lymphoma [12, 13].
In a prospective study of patients with WM, treated with a nucleoside analog or an alkylating agent, transformation was reported in approximately 8-11% at 6 years [14]. The 15-year probability of developing transformation, after nucleoside analogues for WM, was reported as 21% in another series [10]. However, the cumulative incidence of transformation ranged from 2.4% at 10 years to 3.8% at 15 years in a retrospective study of untreated and treated WM patients [15]. Transformation in WM is suggested to occur in less than 10% of patients [10, 13, 15, 16]. About one-quarter of HT has been reported in treatment-naive patients [13, 15].
HT to DLBCL can vary from 1 year to greater than 10 years from diagnosis of WM with a median of 4 - 6 years. Transformation presents rapidly with constitutional B symptoms, splenomegaly and lymphadenopathy as well as rising LDH levels [13, 15, 16]. No guidelines exist on how to screen patients at risk of HT in WM. Diagnosis is made with definitive biopsy. A nongerminal cell DLBCL phenotype is the common transformed subtype [10, 13, 15, 16].
PET-CT avidity, used to suggest transformation, ranged in standardized uptake value (SUV) levels from 3.3 to 38 with median average of 7 - 15 [13, 17]. Extranodal involvement, particularly MYD88-associated sites, such as central nervous system (CNS), skin and testis have been reported at the time of HT with bone marrow commonly involved [10, 13, 15, 16]. The IgM level can decline at the time of HT attributed to clonal dedifferentiation [13].
Factors associated with HT of WM are not well understood. Prior treatment has shown associations for transformation although an increased risk with nucleoside analogs has not been borne out [10, 14]. Elevated LDH and time to transformation > 5 years appear as unfavorable features on multivariate analysis [13]. MYD88 wild type in WM has been shown to be an independent predictive factor for HT [18, 19]. Sex, age > 70 years, performance status, serum IgM level, previous exposure to purine analogues or to alkylating agents, hemoglobin < 10 g/dL, platelet count < 100 × 103/µL, WBC < 4 × 103/µL and Ann Arbor stage were not statistically significant in univariate analysis for progression-free survival and overall survival in a case series of 77 patients [13]. While Epstein-Barr virus (EBV) is implicated in the pathophysiology of NHL, case series of transformed WM to DLBCL found that the majority of transformed cases were negative for EBV [13, 15, 16]. Limited molecular testing has revealed heterogeneity [20]. Transformations with clonally related and unrelated DLBCL have both been reported [21-23]. Transformation has been associated with complex cytogenetics [24]. Deletion of chromosome 6q and trisomy 4, as well as deletion of chromosome 17p, has been reported [13, 24]. The MYD88 mutation has been detected in the evolved lymphoid large cell as well as MYC rearrangement [13, 15, 16].
The overall prognosis for WM patients with associated HT to DLBCL is poor. Survival rates range from 5% to 8%, with a median survival of less than 3 years [15, 16]. Transformed WM, diagnosed greater than 5 years, has a reported survival period of 8 - 9 months [13]. The median survival in Richter’s syndrome, synonymous for HT in chronic lymphoid leukemia, is comparably low at 1.1 years [25].
First-line treatment for transformed WM commonly employs conventional CHOP chemotherapy with reported response rate of 61%, while second-line treatments have response rates reported at 48% [13]. DHAP, GEMOX, ICE and MINE have been used [10, 13, 16, 26-28]. For patients that could proceed to HSCT, a 3-year estimated cumulative survival has been reported to be greater than 67% in studies that included transformed WM [13, 26-28].
Whether novel agents as Bruton tyrosine kinase inhibitors and anti-Bcl-2 inhibitors with response rates upwards of 90% in initial treatment of WM can alter the biology and risk for HT remains unknown [15]. Immunotherapy with chimeric antigen receptor T cell has been tried [29, 30].
Conclusion
This case highlights that after a decade of continued stability in WM, not requiring treatment, an acute change in laboratory data with minimally progressive IgM levels, in the absence of B symptoms and clinical findings, may be the harbinger of transformation and at the time of diagnosis can have a rapidly deteriorating clinical course. In this case, the tripling of the LDH as the primary drastic change demonstrates the importance of the rapid increase in LDH as a singly reliable marker for HT. Although there is no early intervention to prevent HT in WM, continued vigilance is warranted. Late transformation has been borne out as a negative variable as the generally indolent course of WM is curtailed with the poor outcome in HT. Although MYD88 wild type is a possible predictive factor for transformation, it is unclear if late transformation is clonally or non-clonally related and further molecular investigation is needed.
Learning points
1) Late HT in untreated WM, although uncommon, has been borne out as a negative variable once diagnosed.
2) An acute change from long-term stability of laboratory data or imaging in untreated WM with minimally progressive IgM levels may be the harbinger of transformation.
3) MYD88 wild type has been associated with risk for HT.
4) LDH can be a single reliable marker for HT.
5) Late transformation can be clonally or non-clonally related and further molecular investigation is needed.
6) Chemoimmunotherapy has been the mainstay for transformed WM followed by allogeneic HSCT if chemosensitive.
7) Future data on novel treatments for WM are needed to determine effect on risk and outcome of HT.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Consent was obtained from the patient.
Author Contributions
EBE is a major contributor in writing the manuscript, figure design and data interpretation. SS and NB participated in writing and editing. MD provided the histologic images and their interpretation. CF is a major contributor in the design and writing of the manuscript and creation of the figure. All authors read and approved the final manuscript.
Data Availability
The data that support the findings of this study are available from the electronic medical records of the patient. Data are available from the authors upon reasonable request and with permission of the patient.
| References | ▴Top |
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
doi pubmed - Paludo J, Ansell SM. Waldenstrom macroglobulinemia: biology, genetics, and therapy. Blood Lymphat Cancer. 2016;6:49-58.
doi pubmed - Bartl R, Frisch B, Mahl G, Burkhardt R, Fateh-Moghadam A, Pappenberger R, Sommerfeld W, et al. Bone marrow histology in Waldenstrom's macroglobulinaemia. Clinical relevance of subtype recognition. Scand J Haematol. 1983;31(4):359-375.
doi pubmed - Harris NL, Bhan AK. B-cell neoplasms of the lymphocytic, lymphoplasmacytoid, and plasma cell types: immunohistologic analysis and clinical correlation. Hum Pathol. 1985;16(8):829-837.
doi - Castillo JJ, Olszewski AJ, Kanan S, Meid K, Hunter ZR, Treon SP. Overall survival and competing risks of death in patients with Waldenstrom macroglobulinaemia: an analysis of the Surveillance, Epidemiology and End Results database. Br J Haematol. 2015;169(1):81-89.
doi pubmed - Dalal NH, Dores GM, Curtis RE, Linet MS, Morton LM. Cause-specific mortality in individuals with lymphoplasmacytic lymphoma/Waldenstrom macroglobulinaemia, 2000-2016. Br J Haematol. 2020;189(6):1107-1118.
doi pubmed - Kapoor P, Ansell SM, Fonseca R, Chanan-Khan A, Kyle RA, Kumar SK, Mikhael JR, et al. Diagnosis and management of Waldenstrom macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) Guidelines 2016. JAMA Oncol. 2017;3(9):1257-1265.
doi pubmed - Wood TA, Frenkel EP. An unusual case of macroglobulinemia. Arch Intern Med. 1967;119(6):631-637.
doi - McCallister BD, Bayrd ED, Harrison EB. Primary macroglobulinemia: review with a report on thirty-one cases and notes on the values of continuous Chlorambucil therapy. American Journal of Medicine. 1967;43(3):394-434.
doi - Leleu X, Soumerai J, Roccaro A, Hatjiharissi E, Hunter ZR, Manning R, Ciccarelli BT, et al. Increased incidence of transformation and myelodysplasia/acute leukemia in patients with Waldenstrom macroglobulinemia treated with nucleoside analogs. J Clin Oncol. 2009;27(2):250-255.
doi pubmed - Owen RG, Bynoe AG, Varghese A, de Tute RM, Rawstron AC. Heterogeneity of histological transformation events in Waldenstrom's macroglobulinemia (WM) and related disorders. Clin Lymphoma Myeloma Leuk. 2011;11(1):176-179.
doi pubmed - Rosales CM, Lin P, Mansoor A, Bueso-Ramos C, Medeiros LJ. Lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia associated with Hodgkin disease. A report of two cases. Am J Clin Pathol. 2001;116(1):34-40.
doi pubmed - Durot E, Tomowiak C, Michallet AS, Dupuis J, Hivert B, Lepretre S, Toussaint E, et al. Transformed Waldenstrom macroglobulinaemia: clinical presentation and outcome. A multi-institutional retrospective study of 77 cases from the French Innovative Leukemia Organization (FILO). Br J Haematol. 2017;179(3):439-448.
doi pubmed - Leblond V, Johnson S, Chevret S, Copplestone A, Rule S, Tournilhac O, Seymour JF, et al. Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenstrom macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J Clin Oncol. 2013;31(3):301-307.
doi pubmed - Castillo JJ, Gustine J, Meid K, Dubeau T, Hunter ZR, Treon SP. Histological transformation to diffuse large B-cell lymphoma in patients with Waldenstrom macroglobulinemia. Am J Hematol. 2016;91(10):1032-1035.
doi pubmed - Lin P, Mansoor A, Bueso-Ramos C, Hao S, Lai R, Medeiros LJ. Diffuse large B-cell lymphoma occurring in patients with lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia. Clinicopathologic features of 12 cases. Am J Clin Pathol. 2003;120(2):246-253.
doi pubmed - Banwait R, O'Regan K, Campigotto F, Harris B, Yarar D, Bagshaw M, Leleu X, et al. The role of 18F-FDG PET/CT imaging in Waldenstrom macroglobulinemia. Am J Hematol. 2011;86(7):567-572.
doi pubmed - Zanwar S, Abeykoon JP, Durot E, King R, Perez Burbano GE, Kumar S, Gertz MA, et al. Impact of MYD88(L265P) mutation status on histological transformation of Waldenstrom Macroglobulinemia. Am J Hematol. 2020;95(3):274-281.
doi pubmed - Treon SP, Gustine J, Xu L, Manning RJ, Tsakmaklis N, Demos M, Meid K, et al. MYD88 wild-type Waldenstrom Macroglobulinaemia: differential diagnosis, risk of histological transformation, and overall survival. Br J Haematol. 2018;180(3):374-380.
doi pubmed - Jimenez C, Alonso-Alvarez S, Alcoceba M, Ordonez GR, Garcia-Alvarez M, Prieto-Conde MI, Chillon MC, et al. From Waldenstrom's macroglobulinemia to aggressive diffuse large B-cell lymphoma: a whole-exome analysis of abnormalities leading to transformation. Blood Cancer J. 2017;7(8):e591.
doi pubmed - Shiseki M, Masuda A, Watanabe N, Fujii M, Kimura T, Yoshinaga K, Mori N, et al. Development of diffuse large B-cell lymphoma in a patient with Waldenstrom's macroglobulinemia/lymphoplasmacytic lymphoma: clonal identity between two B-cell neoplasms. Hematol Rep. 2011;3(2):e10.
doi pubmed - Shimizu S, Tamagawa Y, Kojima H, Mori N, Nagata M, Noguchi M, Nagasawa T. Simultaneous development of lymphoplasmacytic lymphoma and diffuse large B-cell lymphoma—analyses of the clonal relatedness by sequencing CDR3 in immunoglobulin heavy chain genes. Eur J Haematol. 2003;70(2):119-124.
doi pubmed - Talaulikar D, Biscoe A, Lim JH, Gibson J, Arthur C, Mackinlay N, Saxena K, et al. Genetic analysis of Diffuse Large B-cell Lymphoma occurring in cases with antecedent Waldenstrom Macroglobulinaemia reveals different patterns of clonal evolution. Br J Haematol. 2019;185(4):767-770.
doi pubmed - Mansoor A, Medeiros LJ, Weber DM, Alexanian R, Hayes K, Jones D, Lai R, et al. Cytogenetic findings in lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia. Chromosomal abnormalities are associated with the polymorphous subtype and an aggressive clinical course. Am J Clin Pathol. 2001;116(4):543-549.
doi pubmed - Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood. 2014;123(11):1647-1657.
doi pubmed - Ban-Hoefen M, Vanderplas A, Crosby-Thompson AL, Abel GA, Czuczman MS, Gordon LI, Kaminski MS, et al. Transformed non-Hodgkin lymphoma in the rituximab era: analysis of the NCCN outcomes database. Br J Haematol. 2013;163(4):487-495.
doi pubmed - Villa D, George A, Seymour JF, Toze CL, Crump M, Lee C, Buckstein R, et al. Favorable outcomes from allogeneic and autologous stem cell transplantation for patients with transformed nonfollicular indolent lymphoma. Biol Blood Marrow Transplant. 2014;20(11):1813-1818.
doi pubmed - Kuruvilla J, MacDonald DA, Kouroukis CT, Cheung M, Olney HJ, Turner AR, Anglin P, et al. Salvage chemotherapy and autologous stem cell transplantation for transformed indolent lymphoma: a subset analysis of NCIC CTG LY12. Blood. 2015;126(6):733-738.
doi pubmed - Vaxman I, Gertz M. Waldenstrom's macroglobulinemia in the era of immunotherapy. Leuk Lymphoma. 2020;61(6):1292-1304.
doi pubmed - Bansal R, Jurcic JG, Sawas A, Mapara MY, Reshef R. Chimeric antigen receptor T cells for treatment of transformed Waldenstrom macroglobulinemia. Leuk Lymphoma. 2020;61(2):465-468.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.