

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Original Article
Volume 8, Number 3, September 2019, pages 89-101
Changes in IDH2, TET2 and KDM2B Gene Expression After Treatment With Classic Chemotherapeutic Agents and Decitabine in Myelogenous Leukemia Cell Lines
Jayse Alvesa, Georgia Muccillo Dexheimera, Laura Reckzigelb, Marcia Goetterta, Vanderlei Biolchib, Ana Lucia Abujamraa, c
aGraduate Program in Biotechnology, Univates, Lajeado, R.S., 95914-014, Brazil
bBiological and Health Sciences Center, Univates, Lajeado, R.S., 95914-014, Brazil
cCorresponding Author: Ana Lucia Abujamra, Programa de Pos-Graduacao em Biotecnologia-Univates, Rua Avelino Tallini, 171, Lajeado, RS, 95914-014, Brazil
Manuscript submitted July 9, 2019, accepted August 22, 2019
Short title: Gene Expression in Myelogenous Leukemia
doi: https://doi.org/10.14740/jh531
| Abstract | ▴Top |
Background: Hematological malignancies are a heterogeneous group of tumors with increased proliferative and auto-replicative capacity. Despite treatment advances, post-treatment quality of life remains highly affected. Studies addressing the molecular mechanisms of these diseases are critical for the development of effective, rapid and selective therapies, since few therapeutic strategies succeed in being effective without triggering high-grade toxicities or debilitating late effects. Our aim of this study was to verify changes in the expression of genes involved in the malignant phenotype of hematological malignancies, by treating human cell lines in vitro with classic chemotherapeutic agents and the demethylating agent, decitabine.
Methods: KASUMI-1 and K-562 human myeloid leukemia cell lines were plated at a density of 3 × 104 cells/well and treated with increasing concentrations of different chemotherapeutic agents commonly used in the clinical setting. After 24 and 48 h of treatment, cell viability was tested, and RNA was extracted. Complementary DNA (cDNA) was synthesized and quantitative real-time polymerase chain reaction (qPCR) was performed to evaluate the gene expression of IDH2, TET2 and KDM2B.
Results: A modulation in gene expression was observed before and after treatment with classic chemotherapeutic agents. It was possible to demonstrate a difference in gene expression when cells were treated with chemotherapeutic agents or decitabine alone when compared to chemotherapeutic agents in association with decitabine.
Conclusions: The genes tested, and the modulation of their expression during in vitro treatments suggest that IDH2, TET2, and KDM2B should be further investigated as potential biomarkers for ongoing treatment response and follow-up for patients diagnosed with hematological malignancies of the myeloid lineage.
Keywords: Myeloid leukemia; Gene expression biomarkers; Chemotherapy; Decitabine
| Introduction | ▴Top |
Acute myeloid leukemia (AML) is a hematological malignancy characterized by uncontrolled proliferation of myeloblasts that accumulate in the bone marrow and peripheral blood. This proliferation leads to high levels of malignant and immature leukocytes, and to an abnormal production of platelets and erythrocytes [1], culminating in anemia, thrombocytopenia, and neutropenia [2].
Typically, treatment is divided into three phases: induction to remission, consolidation, and maintenance. In induction to remission, cytotoxic chemotherapy is employed to eliminate circulating and bone marrow leukemic cells until complete remission of the disease is achieved. During consolidation therapy, if a bone marrow transplant is deemed unnecessary, treatment is similar or less intense than the induction therapy, aiming at eliminating any residual malignant cells. Maintenance therapy varies according to the patients’ overall well-being and age [1].
The standard therapy includes 3 days of combined anthracycline treatment with 7 days of cytarabine treatment (Ara-C) [2, 3], aiming to achieve complete remission and prolongation of patient survival. About 70% of patients under 60 years of age achieve complete remission when using this combination [4]; however, older patients and those with comorbidities are often considered ineligible for standard induction therapy, and their treatment options are guided by disease biology, performance status, and clinical limitations, which hinder the efficacy of therapy, or the patient’s ability to tolerate the toxic side effects of induction therapy [2, 3].
Another hematological malignancy with predominance in patients over 60 years of age is chronic myeloid leukemia (CML), responsible for up to 20% of all cases of newly diagnosed leukemia in adults [5]. It is characterized by the excessive proliferation of cells from the myeloid lineage, especially granulocytes in the bone marrow [6] and by the accumulation of these cells in peripheral blood [7].
The main treatment is based on the use of tyrosine kinase inhibitors (TKIs), such as imatinib, for patients holding the reciprocal translocation t(9;22)(q34; q11) [8, 9]. TKIs can achieve durable cytogenetic and molecular remissions, improve survival of most patients, and demonstrate a reasonable disease remission rate, since a high percentage of patients achieve a good molecular response [10]. Although the side effects with TKIs are less frequent and milder than other chemotherapeutic agents, they still require frequent patient follow-ups. Moreover, resistance to TKIs and important treatment side-effects have already been reported in the literature [11-13].
The carcinogenic process initiates at the molecular level, with gene mutations and epigenetic alterations in key genes which participate in cell cycle regulation and proliferation. These changes are used as targets for therapies and markers for diagnosis, prognosis, and treatment response evaluation [14, 15]. Genes such as IDH2 (isocitrate dehydrogenase 2), TET2 (Ten Eleven Translocation 2), and KDM2B (lysine demethylase 2B) are considered epigenetic regulators [16]. Changes in expression of IDH2 and TET2 have already been related to decreased myeloid differentiation patterns [17]. The KDM2B gene is a histone demethylase that favors oncogenesis in the lymphoid line and suppresses oncogenesis the myeloid lineage [18].
Decitabine is a potent demethylating agent capable of inducing hypomethylation, demonstrating the ability of reactivating several genes. It is used for the treatment of myelodysplastic syndrome (MDS) and AML, with promising, but not fully curative, results. Presently, decitabine shows limitations in about half of the patients treated, being that less than half of the patients treated with this drug achieve full remission [19, 20]. Clinical studies have shown that the combination of classic therapeutic regimens and decitabine may yield satisfactory results [21, 22]. Moreover, at the present date, there are 58 ongoing clinical trials for different types of leukemia using decitabine combined with other drugs [23].
AML and CML are dynamic diseases with established treatments according to the characteristics found during the diagnostic process. However, the disease may present changes over time and adaptations to treatment are necessary for treatment success. In this way, studies that can identify tumor adaptations and/or modifications are important for disease control [24]. The aim of this study was to verify whether treatment with classic chemotherapeutic agents, decitabine, or a combination of both was able to modify the expression of IDH2, TET2 and KDM2B in two commercially available human myelogenous leukemia cell lines.
| Materials and Methods | ▴Top |
Cell lines and reagents
American Type Culture Collection (ATCC) AML (KASUMI-1) and CML (K-562) human cell lines were obtained via a donation from the Laboratory of Bone Marrow and Stem Cells of the Federal University of Sso Paulo, UNIFESP. Cells were grown in 25 cm2 flasks containing 7 mL of RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS, Nutricell) and 1% antifungal/antibiotic solution (Sigma-Aldrich; St. Louis, USA) and maintained at 37 °C in a humidified incubator with 5% atmospheric CO2. Cytarabine (Ara-C) and methotrexate (MTX) (Libbs; Sao Paulo, Brazil); etoposide (VP-16) (Blau Farmaceutica; Cotia, Brazil) and vincristine (VCR) (Zodiac; Pindamonhangaba, Brazil) were donated by Oncosinos, located in Sao Leopoldo, Brazil. Doxorubicin (Doxo) was obtained from Sigma-Aldrich (St. Louis, USA) and decitabine (Deci) from Caymann (Ann Arbor, USA).
Evaluation of cellular proliferation
Cells were seeded in 4- and 24-well plates suitable for growth of cells in suspension at a density of 3 × 104 cells/well. Cells were then treated with high (IC70) or half maximal inhibitory concentration (IC50) doses of Ara-C (25 µM or 10 µM), MTX (100 µM or 50 µM), VP-16 (25 µM or 1 µM), VCR (2.5 µM or 1 µM), Doxo (25 µM or 1 µM) or Deci 100 nM. Cells were also treated with a combination of each chemotherapeutic agent in its IC50 dose with Deci 100 nM. Twenty-four and 48 h later, 10 µL of cellular suspension was homogenized 1:1 with 0.4% trypan blue solution (Sigma-Aldrich). Cells were counted in a hemocytometer and viability was determined by the trypan blue exclusion test. Results are expressed as absolute cell counts.
Total RNA extraction
Forty-eight hours post-treatment, after cells were counted, the culture medium containing cells in suspension was removed from each well and centrifuged for 10 min at 1,000 g. The supernatant was discarded and RNA from each cell pellet was extracted using the Trizol™ reagent (Invitrogen), according to the manufacturer’s instructions. The final RNA pellet was resuspended in water previously treated with diethylpyrocarbonate (DEPC).
Complementary DNA (cDNA) synthesis
cDNA synthesis was performed using the SuperScript™ First-Strand Synthesis System for qPCR kit (Thermo Fisher) utilizing oligodT primers, according to the manufacturer’s instructions. Briefly, 500 ng of RNA was utilized for a reaction containing 1 µL 10 mM deoxyribonucleoside triphosphates (dNTPs), 50 µM oligo (dT)20, and DEPC-treatd water in a final reaction volume of 10 µL. After a 5-min incubation period at 65 °C, 10 µL buffer containing 25 mM MgCl2, 0.1 M DTT, 1 µL RNAseOUT™ and 1 µL SuperScript III RT™ was added. Samples were then incubated at 50 °C for 50 min for synthesis, which was terminated at 85 °C for 5 min. A sample included with the kit was used as a positive control, and a sample without RNA was included as a negative control.
Real-time PCR
Quantitative real-time PCR reactions were performed in a StepOne Real Time PCR System (AppliedBiosystems), with the primers described in Table 1 and utilizing the conditions described in Tables 2 and 3, for K562 and KASUMI-1 cell lines, respectively. Data were analyzed using the comparative Ct method. PCR efficiency from the exponential phase was calculated for each individual amplification plot with the LinRegPCR software version 11.0. In each plate, the average of PCR efficiency (Eff) for each amplicon was determined and used in future calculations. Ct values of β-actin and GAPDH were used to normalize Ct values for each gene tested. Gene expression was calculated based on the ratio to β-actin and GAPDH. Each data point corresponds to three true biological replicate samples.
 Click to view | Table 1. Forward and Reverse Primers Used for Gene Amplification |
Click to view | Table 2. Standardization of qPCR Reaction for IDH2, TET2 and KDM2B Genes in the K-562 Cell Line |
Click to view | Table 3. Standardization of qPCR Reaction for IDH2, TET2 and KDM2B Genes in the KASUMI-1 Cell Line |
Statistical analysis
Data were analyzed with descriptive statistics using SPSS software (Statistical Package of Social Sciences, IBM Inc.) version 19.0. Normality tests were applied to verify whether data distribution was parametric or non-parametric. If parametric, the results were expressed as mean ± standard deviation (SD) of the mean. If non-parametric, results were expressed as median and their interquartile ranges (25% and 75%). For parametric results, further analysis was then carried out via one-way ANOVA, followed by the Tukey post-hoc test to verify if there were differences between groups. For non-parametric results, a Kruskal-Wallis test was employed, followed by the Dunn’s multiple correlation test. The significance level adopted was 5%, with values of P < 0.05 being considered significant.
The work described here does not contain any studies with human participants or animals.
| Results | ▴Top |
After establishing cell growth curves and optimizing cellular density for each plate, dose curves were performed for each chemotherapeutic agent and for decitabine in the KASUMI-1 and K-562 cell lines to verify each reagent’s cytotoxic activity. The chemotherapeutic doses were selected according to the IC50 and IC70 for each agent. IC50 concentrations for each chemotherapeutic agent were: Doxo 1 µM, VP-16 1 µM, VCR 1 µM, Ara-C 10 µM, and MTX 50 µM. Decitabine is considered a demethylating agent and showed a cytotoxic effect on both cell lines tested, reducing cellular proliferation in a dose-dependent manner (Fig. 1). For the combination treatments, Deci was employed at 100 nM, which is a concentration that does not affect cellular proliferation significantly (data not shown).
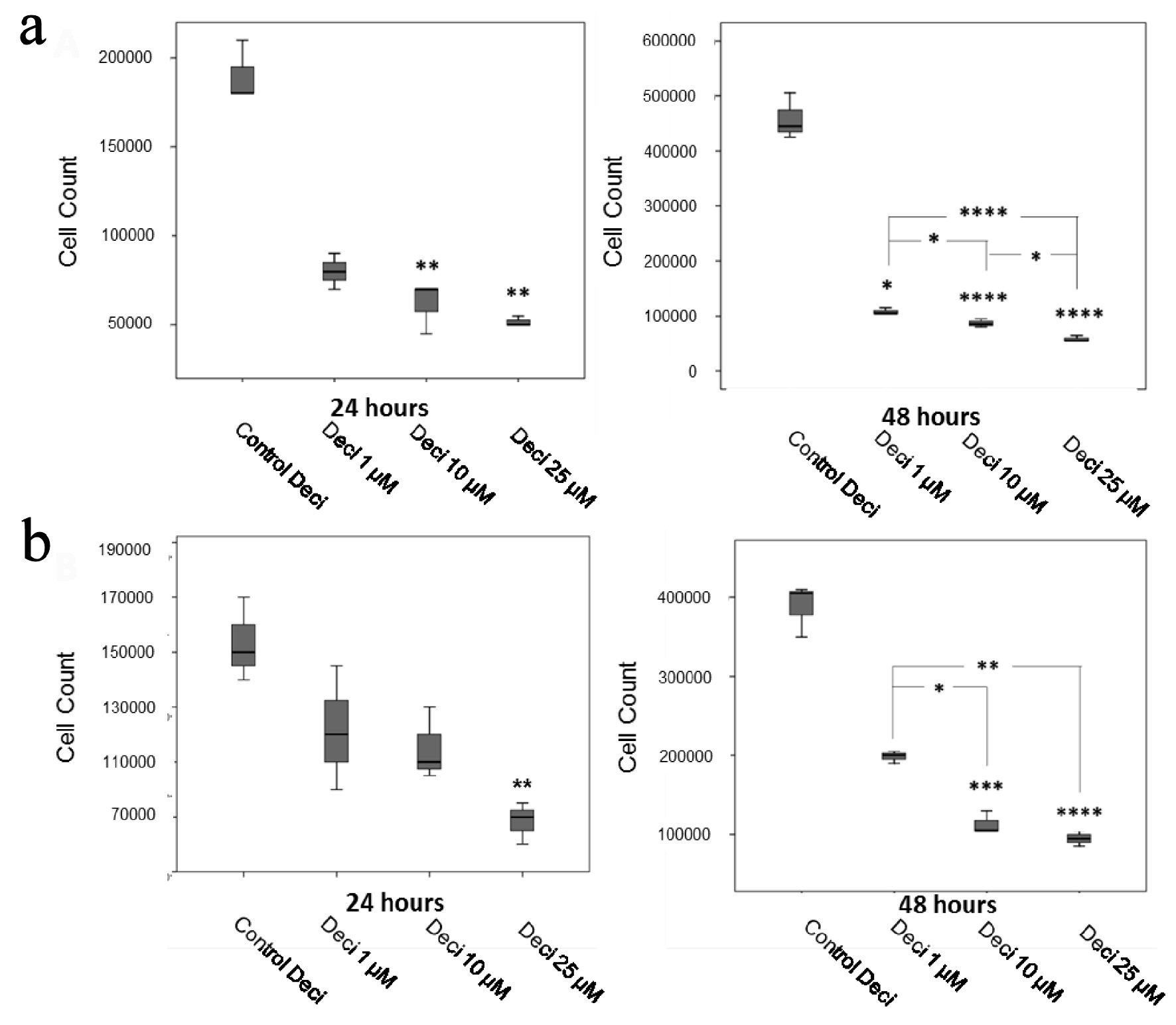 Click for large image | Figure 1. Decitabine dose curves in the KASUMI-1 cell line (a) and in the K-562 cell line (b). P values were determined using one-way ANOVA, followed by the Tukey post-hoc test (parametric), or using Kruskal-Wallis test, followed by Dunn’s multiple correlations test (non-parametric), using P < 0.05 for statistically significant differences. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Results are expressed as absolute cell counts. |
With regards to the KASUMI-1 cell line, all chemotherapeutic agents tested at the IC70 and IC50 concentrations yielded a significant decrease in cellular proliferation when compared to decitabine alone after 48 h post-treatment. No differences were found in cellular proliferation when comparing chemotherapeutic agents alone and associated with Deci 100 nM (Fig. 2). In the K-562 cell line, all chemotherapeutic agents tested at the IC70 and IC50 concentrations yielded a significant decrease in cellular proliferation when compared to decitabine alone after 24- and 48-h post-treatment (Fig. 3). When comparing each chemotherapeutic agent alone as well as associated with decitabine, there were significant differences only in the treatments with etoposide (P < 0.05) (Fig. 3b) and vincristine (P < 0.0001) (Fig. 3c).
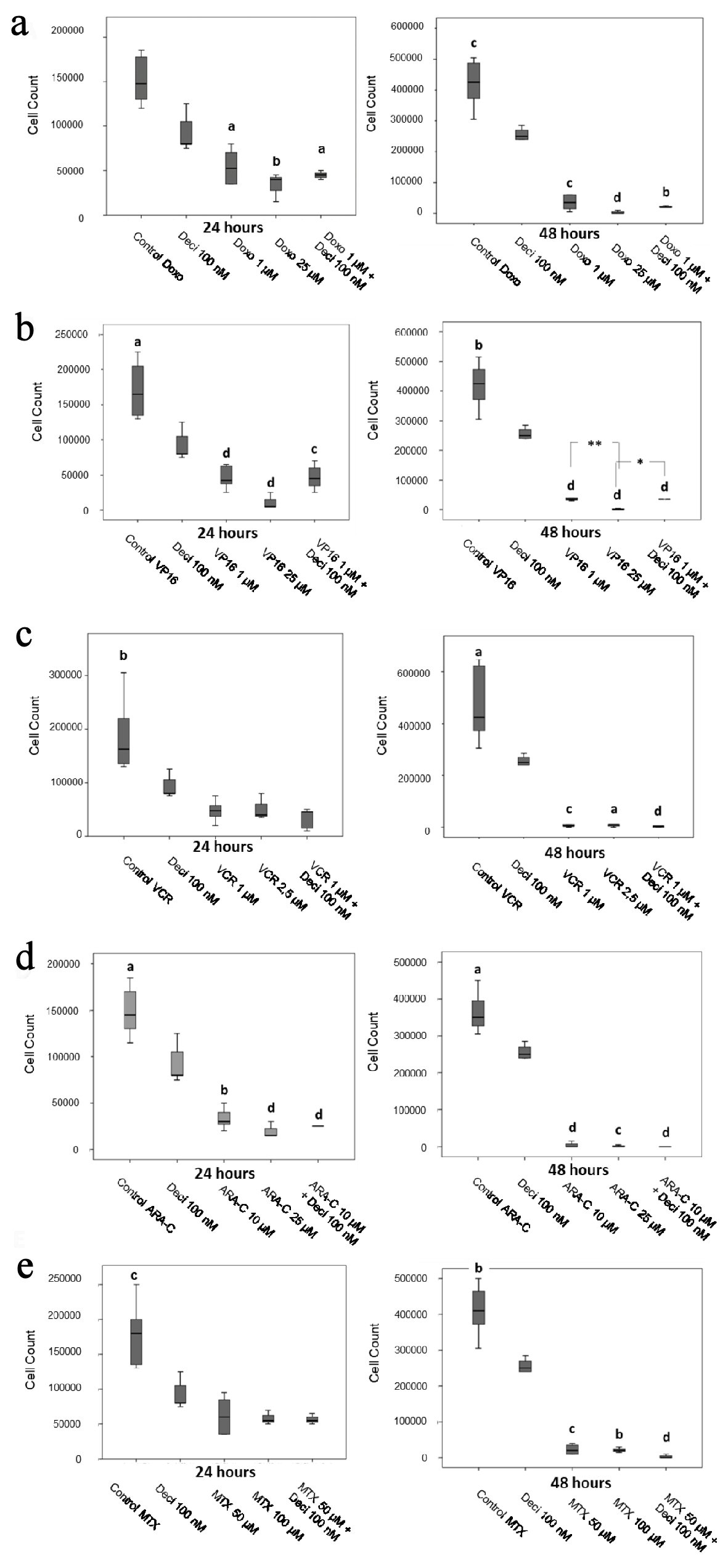 Click for large image | Figure 2. Cell counts after 24 and 48 h of treatment with doxorubicin (a), etoposide (b), vincristine (c), cytarabine (d) and methotrexate (e) in the KASUMI-1 cell line alone or in association with decitabine. The differences shown are in relation to treatment with decitabine. Values were determined using one-way ANOVA, followed by the Tukey post-hoc test (parametric), or Kruskal-Wallis test, followed by Dunn’s multiple correlations test (non-parametric), where P < 0.05 values determined statistically significant differences. a: P < 0.05, b: P < 0.01, c: P < 0.001, d: P < 0.0001. Results are expressed as absolute cell counts. |
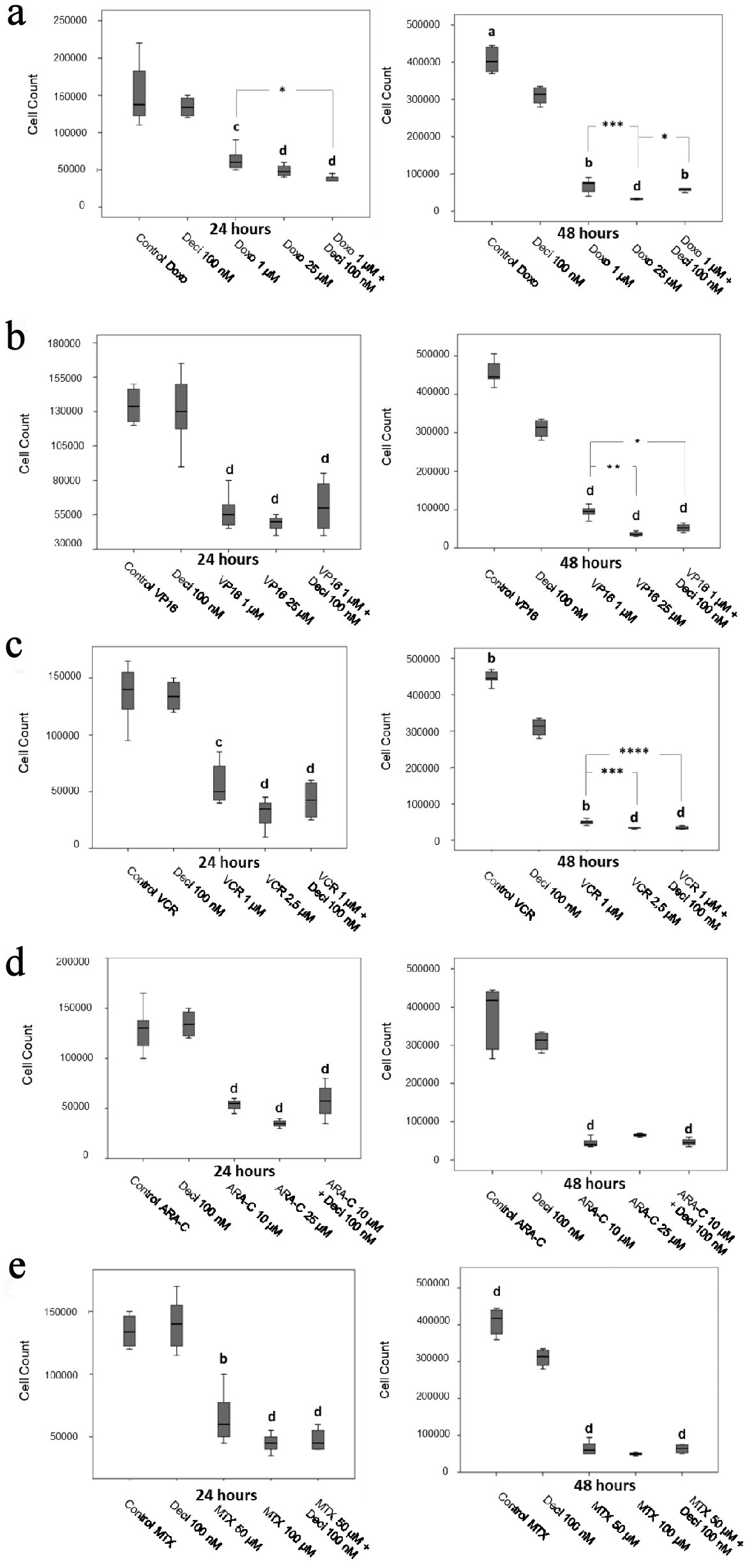 Click for large image | Figure 3. Cell counts after 24 and 48 h of treatment with doxorubicin (a), etoposide (b), vincristine (c), cytarabine (d) and methotrexate (e) in the K-562 cell line. The differences shown are in relation to treatment with decitabine. P values were determined using the Kruskal-Wallis test, followed by Dunn’s multiple correlation test, using P < 0.05 to determine statistically significant differences. */a: P < 0.05, b: P < 0.01, c/***: P < 0.001, d: P < 0.0001; values of P represented in letters are relative to decitabine; values of P represented in * are relative to the isolated chemotherapeutic treatment. Results are expressed as absolute cell counts. |
Gene expression levels of TET2, IDH2 and KDM2B in both untreated cell lines were compared to expression levels of TET2, IDH2 and KDM2B after treatment with each chemotherapeutic agent alone, decitabine alone, or an association of both in order to verify whether any of these treatments could modulate gene expression. IDH2 gene expression in the KASUMI-1 cell line was significantly elevated (P < 0.0001 for all combinations) when treated with a combination of chemotherapeutic agents in association with decitabine, when compared to chemotherapy alone (Fig. 4), except in the case of vincristine (Fig. 4d). In the K-562 cell line, IDH2 gene expression increased after treatment with an association of decitabine and etoposide (P < 0.001) (Fig. 4g), vincristine (P < 0.0001) (Fig. 4h), and methotrexate (P < 0.01) (Fig. 4j) when compared to each chemotherapeutic agent alone.
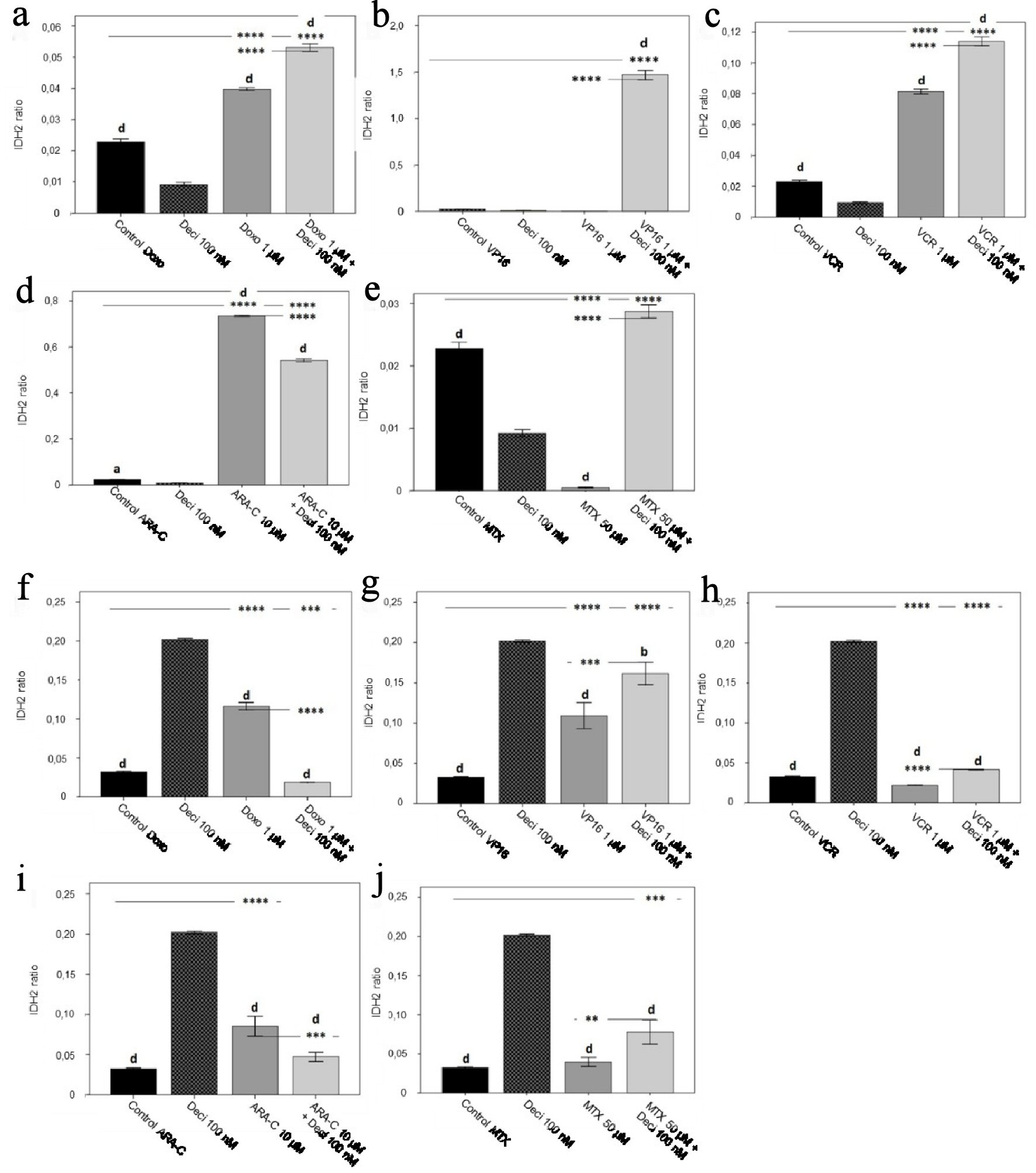 Click for large image | Figure 4. IDH2 gene expression levels in the KASUMI-1 cell line after treatment with (a) doxorubicin, (b) etoposide, (c) vincristine, (d) cytarabine, and (e) methotrexate, and in the K-562 cell line after treatment with (f) doxorubicin, (g) etoposide, (h) vincristine, (i) cytarabine, and (j) methotrexate. The differences shown are in relation to treatment with decitabine. P values were determined using one-way ANOVA, followed by the Tukey post-hoc test, using P < 0.05 to determine statistically significant differences. a/*: P < 0.05, b/**: P < 0.01, c/***: P < 0.001, d/****: P < 0.0001; values of P represented in letters are relative to decitabine; values of P represented in * are relative to the isolated control or chemotherapeutic agent. |
Gene expression of KDM2B in the KASUMI-1 cell line was significantly increased (P < 0.0001) after combined treatment of decitabine and doxorubicin (Fig. 5a), etoposide (Fig. 5b) and cytarabine (Fig. 5d) when compared to each chemotherapeutic agent alone. Treatment with vincristine (Fig. 5c) and methotrexate (Fig. 5e) significantly decreased the expression of KDM2B (P < 0.0001) when in combination with decitabine, as compared to chemotherapy alone. In the K-562 cell line, KDM2B gene expression was significantly decreased after treatment with all chemotherapeutic agents in association with decitabine when compared to chemotherapy alone (Fig. 5), except for etoposide (Fig. 5g), which presented a significant increase (P < 0.0001) in gene expression.
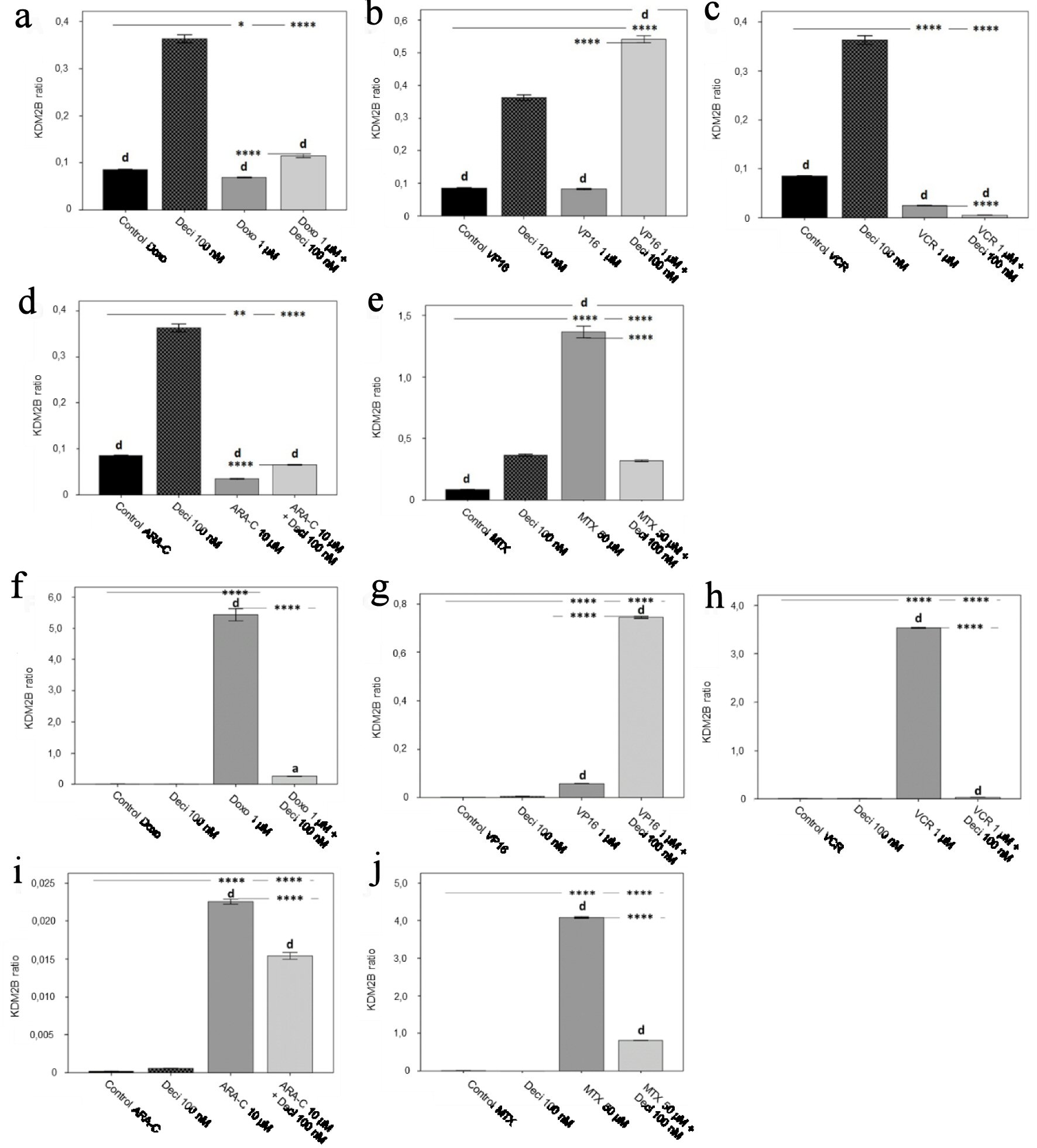 Click for large image | Figure 5. KDM2B gene expression levels in the KASUMI-1 cell line after treatment with (a) doxorubicin, (b) etoposide, (c) vincristine, (d) cytarabine, and (e) methotrexate, and in the K-562 cell line after treatment with (f) doxorubicin, (g) etoposide, (h) vincristine, (i) cytarabine, and (j) methotrexate. The differences shown are in relation to treatment with decitabine. P values were determined using one-way ANOVA, followed by the Tukey post-hoc test, using P < 0.05 to determine statistically significant differences. a/*: P < 0.05, b/**: P < 0.01, c/***: P < 0.001, d/****: P < 0.0001; values of P represented in letters are relative to decitabine; values of P represented in * are relative to the isolated control or chemotherapeutic agent. |
With regards to TET2 gene expression, when comparing each chemotherapeutic agent associated with decitabine versus treatment with chemotherapy alone, a decrease in gene expression (P < 0.0001) was observed for doxorubicin (Fig. 6a), vincristine (Fig. 6c), and methotrexate (Fig. 6e) in the KASUMI-1 cell line. Etoposide (Fig. 6b) and cytarabine (Fig. 6d) treatments increased TET2 gene expression when associated with decitabine as compared to treatment with chemotherapy alone (P < 0.0001). In the K-562 cell line, treatment with doxorubicine (Fig. 6f), etoposide (Fig. 6g), and methotrexate (Fig. 6j) in association with decitabine increased TET2 gene expression when compared to each chemotherapeutic agent alone (P < 0.0001). TET2 gene expression was significantly decreased after treatment with decitabine and vincristine (Fig. 6h) or cytarabine (Fig. 6i) when compared to treatment with vincristine or cytarabine alone (P < 0.0001).
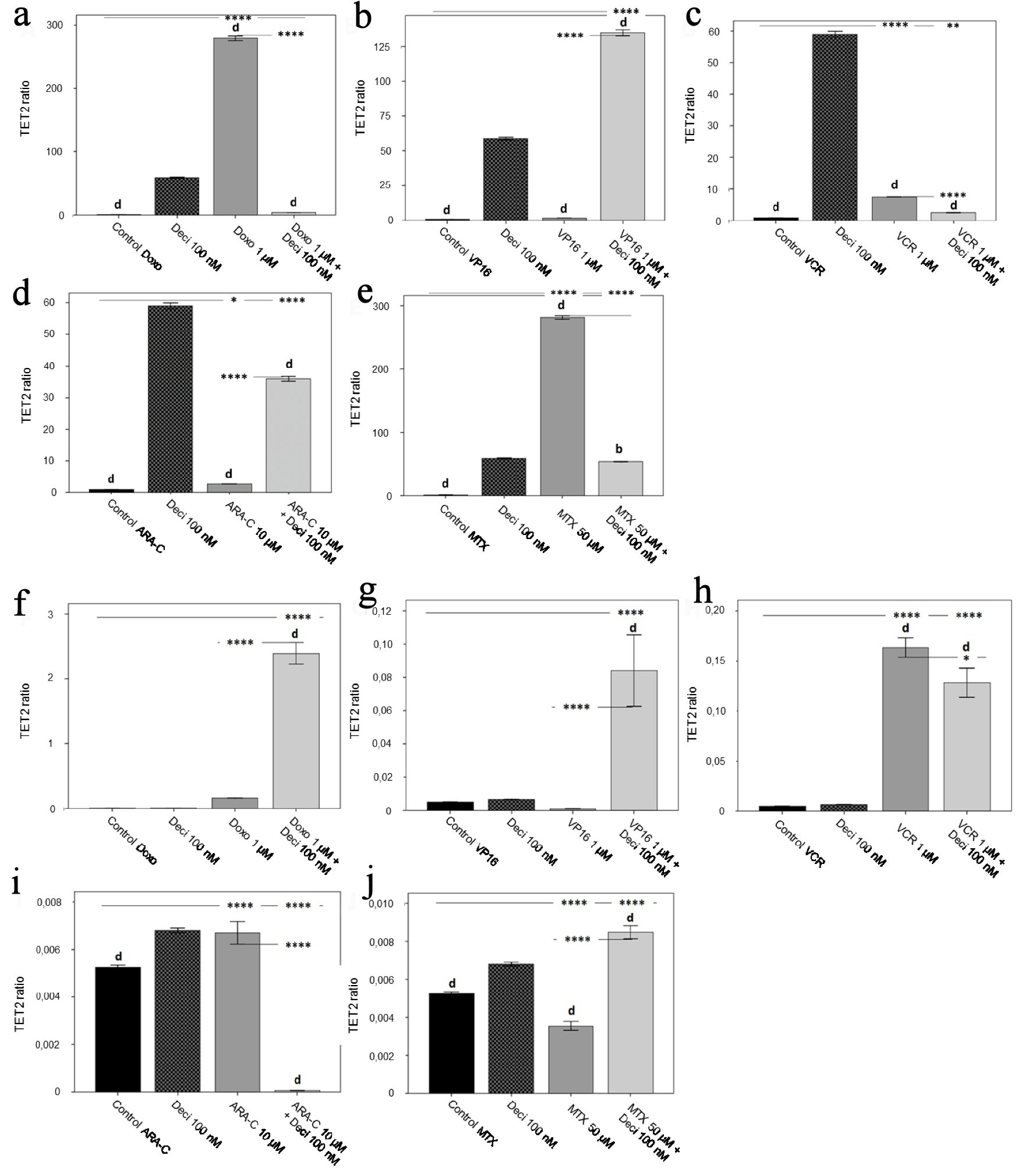 Click for large image | Figure 6. TET2 gene expression level in the KASUMI-1 cell line after treatment with (a) doxorubicin, (b) etoposide, (c) vincristine, (d) cytarabine, and (e) methotrexate, and in the K-562 cell line after treatment with (f) doxorubicin, (g) etoposide, (h) vincristine, (i) cytarabine, and (j) methotrexate. The differences shown are in relation to treatment with decitabine. P values were determined using one-way ANOVA, followed by the Tukey post-hoc test using P < 0.05 to determine statistically significant differences. a/*: P < 0.05, b/**: P < 0.01, c/***: P < 0.001, d/****: P < 0.0001; values of P represented in letters are relative to decitabine; values of P represented in * are relative to the isolated control or chemotherapeutic agent. |
| Discussion | ▴Top |
The present study evaluated the effects of combining decitabine with classical chemotherapeutic agents used in the treatment of different onco-hematological diseases in the expression of epigenetic regulatory genes which play a part in leukemogenesis: IDH2, TET2, and KDM2B. As expected, the chemotherapeutic agents demonstrated cytotoxic effects in both cell lines used (KASUMI-1 and K-562). We aimed at associating low-dose decitabine with intermediate doses of each chemotherapeutic agent in such a way that this association would not alter cellular proliferation. In this way, we would be able to observe whether there were any changes at the molecular level that preceded manifestations at the cellular level. Here we report that the association of decitabine with classical chemotherapeutic agents modulates gene expression of IDH2, TET2 and KDM2B when compared to treatment with each chemotherapeutic agent alone. For the most part, these changes in gene expression occurred all the while not altering cellular proliferation.
Here we also evaluated the expression levels of IDH2, TET2 and KDM2B after treatment with decitabine alone, and compared it to untreated controls, to treatment with each chemotherapeutic agent alone, and to combination treatments of chemotherapy plus low-dose decitabine. These comparisons may indicate in which situations the association may modify the expression of each gene tested, more so than with each drug by itself. It is important to emphasize that decitabine is already employed for the treatment of certain hematological malignancies; however its use has not been proven efficient as monotherapy. For this reason, this discussion will focus on the modulation of gene expression as a result of the association of decitabine with other classical chemotherapeutic agents.
In neoplastic cells, decitabine can reactivate silenced genes and inhibit DNA methylation in low doses; cytotoxic effects have been observed in high doses. Patients with MDS/AML, CML and ALL received different doses of decitabine, ranging 5- to 30-fold lower than the maximum tolerated dose. Responses were observed at all doses studied, however low-dose decitabine was as effective, and, at times, more effective than high-dose decitabine [25]. Kopp and collaborators (2013) [26] showed that decitabine treatment presents a biphasic response with regards to hypomethylation and cytotoxicity. Decitabine affects the expression of activating and inhibitory receptors in natural killer cells at low doses during cellular proliferation. High-dose decitabine can decrease natural killer cell proliferation and reduce viability through inhibition of mRNA transcription. This data corroborate that of Issa and collaborators (2004) [25], and suggests that decitabine’s optimal immunomodulation occurs at low doses, whereas high doses are associated with inhibition of proliferation and direct toxicity to cells. Low doses of decitabine also induced differentiation and maturation of myelodysplastic megakaryocytes in MDS patients, which is useful in the clinical setting [27].
A multicenter analysis performed in patients with intermediate- or high-risk MDS exploring decitabine in very low doses showed reasonable efficacy and tolerability, as well as lower medical costs [28]. Taking into consideration the costs involved in the treatment of oncological diseases, studies demonstrating that low doses of decitabine are equally efficient as higher doses are welcome. This also explains why the association of hypomethylating agents, such as decitabine, with other pharmacological treatments is widely used in the search for more efficient and cost-effective therapies [29-31].
Low doses of decitabine were associated with clinical improvement and hematological remission in MDS/MPN patients [32], disease remission and hematologic improvement in AML and MDS patients [33], and reduction of cell viability and induction of apoptosis in ALL patients [34]. The use of low-dose hypomethylating agents is safe and effective in patients with low-risk MDS and MDS/MPN [35] as well as in high-risk MDS [28]. Moreover, there are favorable data with regards to decitabine’s effects on overall survival [2], toxicity profile and clinical efficacy [36]. These data are in accordance with what was observed in this study: decitabine can greatly modulate gene expression differently than chemotherapeutic agents by themselves. Here, even though changes in gene expression as a result of decitabine treatment did not necessarily translate to changes in cell behavior in vitro, the fact that low doses of decitabine in combination with chemotherapeutic agents modulated the expression of IDH2, TET2 and KDM2B, more so than with each treatment alone, strengthens the argument that decitabine should be considered for combination therapy with the chemotherapeutic agents currently employed in the clinical setting.
IDH2 plays a role in energy production and a crucial role in cellular proliferation [37]. Mutations in IDH2 can lead to decreased myeloid differentiation, and has already been associated with the development of MDS and AML [17, 38, 39]. Its downregulation was found to exacerbate the malignant progression of osteosarcoma cells via NF-kB and MMP9 pathway activation [40]. Leukemic cells showed decreased expression of IDH2 when compared to untransformed B cells; lower IDH2 expression accelerates cell cycle progression and increases the tumor’s invasive capacity, all of which are associated with malignant progression [41]. Here we demonstrate that in the KASUMI-1 cell line the association of decitabine with doxorubicin, etoposide, cytarabine and methotrexate increased IDH2 gene expression. The same increase in IDH2 expression was observed in the K-562 cell line when decitabine was associated with etoposide, vincristine and methotrexate. This suggests that the association of low-dose decitabine with certain chemotherapeutic agents may be beneficial, for it modulates expression of a gene that is crucial for the leukemogenic process.
Recent studies involving theTET2 gene suggests that its loss may be a key to leukemic transformation. It has been shown that this gene is important in normal myelopoiesis and that its dysregulation favors myeloid tumorigenesis [42]. Mice with loss of TET2 showed increased hematopoietic stem cell (HSC) turnover and myeloproliferation, associated with splenomegaly, monocytosis, and extramedullary hematopoiesis [43]. Other studies have also shown that the inactivation of TET2 leads to a decrease of 5-hC in HSCs of the myeloid lineage [44, 45]. Changes in the TET2 gene have already been described as an important event in the pathogenesis of different myeloid malignancies including SMD, SMP and AML [46]. Moreover, TET2 gene expression levels are also significantly reduced in breast, liver, lung, pancreas and prostate tumors [47]. In the present study, we demonstrate that, in the KASUMI-1 cell line, the association of decitabine with etoposide or cytarabine significantly increased TET2 gene expression. The same was seen in the K-562 cell line when decitabine was associated with doxorubicine, etoposide, or methotrexate. Thus, associating low-dose decitabine to an already established therapeutic regimen may increase TET2 expression, which could contribute to a more efficient and long-lasting therapeutic response.
The KDM2B gene exhibits opposing roles in hematological malignancies, as it can promote or antagonize tumor progression depending on the cellular context. This gene is highly expressed in leukemias exhibiting protoncogenic activity via repression of tumor suppressor pathways and is highly expressed in murine and human HSPCs compared to other tissues [18, 48]. KDM2B is required for the maintenance of human leukemia cell lines via the regulation of cell fate and lymphocyte-specific signaling pathways. Analysis of gene expression changes in human leukemia cell lines revealed a pleiotropic role for KDM2B in differentiation, quiescence, and lymphoid lineage specification through modulation of cell signaling; KDM2B also plays an important role in lymphoid commitment and T cell maturation [49, 50]. Here we demonstrate that the association of decitabine with vincristine or methotrexate in the KASUMI-1 cell line, and with doxorubicin, vincristine, cytarabine or methotrexate in the K-562 cell line decreased KDM2B gene expression. This suggests that the association of decitabine with other cytotoxic drugs may improve treatment response via epigenetic modulation. Further studies need to be undertaken in order to better explore how restoring cellular differentiation in leukemia may be employed as a viable therapeutic approach. The investigation of KDM2B modulation may provide further information regarding this matter.
Conclusions
Considering the information already presented with regards to decitabine’s effects on cellular differentiation and epigenetic modulation, it is imperative to investigate how this drug may be employed in a safe and economically viable manner to current treatments that are already in order. Here we demonstrate that decitabine at low doses, when combined with certain chemotherapeutic agents already employed in the clinical setting, can modulate three genes that are key to the leukemogenic process: IDH2, TET2, and KDM2B. Moreover, these changes in gene expression were observed before any changes in cellular proliferation were seen, strengthening the rationale for investigating these genes as potential biomarkers for tumor regression and/or disease relapse. Epigenetic modulation has proven to be a key event in disease remission, and may present other benefits besides potentiation of cellular toxicity, such as cellular differentiation, reduction of adverse events and toxicity to treatment, stability of disease via avoidance of gene switching, and an increase in disease-free survival. This report strengthens the rationale for the use of combined, gene-modulating treatments. Further studies are necessary to fully elucidate the exact mechanisms by which this gene modulation occur, and its long-term effects post-treatment.
Acknowledgments
None to declare.
Financial Disclosure
This work was supported by the Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq; grant number 303567/2016-3 to A.L.A), by the Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior (CAPES; to J.A.), by the Fundacao de Amparo a Pesquisa do Estado do Rio Grande do Sul (FAPERGS; to L.R.) and by Fundacao Vale do Taquari de Educacao e Desenvolvimento Social (FUVATES; grant number 10901246 to A.L.A. and L.R).
Conflict of Interest
The authors hereby declare that there is no conflict of interest pertaining to the work described here.
Informed Consent
Not applicable.
Author Contributions
Study concept and design: ALA. Experiments: JA, GMD and LR. Data analysis and statistics: MG, ALA and VB. Figures: JA and GMD. Manuscript preparation and submission: ALA, JA and GMD.
| References | ▴Top |
- Khwaja A, Bjorkholm M, Gale RE, Levine Ross RL, Craig TD, Ehninger G, Bloomfield CD, et al. Acute myeloid leukaemia. Nat Rev Dis Primers. 2016;2:16010.
doi pubmed - Nieto M, Demolis P, Behanzin E, Moreau A, Hudson I, Flores B, Stemplewski H, et al. The European medicines agency review of decitabine (Dacogen) for the treatment of adult patients with acute myeloid leukemia: summary of the scientific assessment of the committee for medicinal products for human use. Oncologist. 2016;21(6):692-700.
doi pubmed - Hasserjian RP. Acute myeloid leukemia: advances in diagnosis and classification. Int J Lab Hematol. 2013;35(3):358-366.
doi pubmed - Sanz MA, Iacoboni G, Montesinos P, Venditti A. Emerging strategies for the treatment of older patients with acute myeloid leukemia. Ann Hematol. 2016;95(10):1583-1593.
doi pubmed - Soverini S, Branford S, Nicolini FE, Talpaz M, Deininger MW, Martinelli G, Muller MC, et al. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leuk Res. 2014;38(1):10-20.
doi pubmed - Khajapeer KV, Baskaran R. Hsp90 Inhibitors for the Treatment of Chronic Myeloid Leukemia. Leuk Res Treatment. 2015;2015:757694.
doi pubmed - Renzi C, Riva S, Masiero M, Pravettoni G. The choice dilemma in chronic hematological conditions: Why choosing is not only a medical issue? A psycho-cognitive perspective. Crit Rev Oncol Hematol. 2016;99:134-140.
doi pubmed - Eyupoglu D, Bozkurt S, Haznedaroglu I, Buyukasik Y, Guven D. The Impact of Variant Philadelphia Chromosome Translocations on the Clinical Course of Chronic Myeloid Leukemia. Turk J Haematol. 2016;33(1):60-65.
doi pubmed - Hoffbrand AV, Moss PAH, Pettit JE. Fundamentos em hematologia, 5 edicao, Porto Alegre, Artmed (BR). 2006.
- Saussele S, Richter J, Hochhaus A, Mahon FX. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia. 2016;30(8):1638-1647.
doi pubmed - Al-Amri AM. outcome of chronic myeloid leukemia-chronic phase patients treated with imatinib: a local experience. Clin Lymphoma Myeloma Leuk. 2018;18(3):199-203.
doi pubmed - Sorokin M, Kholodenko R, Grekhova A, Suntsova M, Pustovalova M, Vorobyeva N, Kholodenko I, et al. Acquired resistance to tyrosine kinase inhibitors may be linked with the decreased sensitivity to X-ray irradiation. Oncotarget. 2018;9(4):5111-5124.
doi pubmed - Zhang Y, Gao Y, Zhang H, Zhang J, He F, Hnizda A, Qian M, et al. PDGFRB mutation and tyrosine kinase inhibitor resistance in Ph-like acute lymphoblastic leukemia. Blood. 2018;131(20):2256-2261.
doi pubmed - Aithal MG, Rajeswari N. Role of Notch signalling pathway in cancer and its association with DNA methylation. J Genet. 2013;92(3):667-675.
doi - Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81(4):303-311.
doi pubmed - Im AP, Sehgal AR, Carroll MP, Smith BD, Tefferi A, Johnson DE, Boyiadzis M. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. 2014;28(9):1774-1783.
doi pubmed - Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553-567.
doi pubmed - Andricovich J, Kai Y, Peng W, Foudi A, Tzatsos A. Histone demethylase KDM2B regulates lineage commitment in normal and malignant hematopoiesis. J Clin Invest. 2016;126(3):905-920.
doi pubmed - Joeckel TE, Lubbert M. Clinical results with the DNA hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in patients with myelodysplastic syndromes: an update. Semin Hematol. 2012;49(4):330-341.
doi pubmed - Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O'Brien S, Cortes J, Faderl S, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109(1):52-57.
doi pubmed - Wang H, Yang B, Chi X, Cai L, Yu R, Zhu H, Tuo S, et al. Ultra-low-dose decitabine combined with autologous cytokine-induced killer cells for elderly patients with acute myeloid leukemia transformed from myelodysplastic syndrome. Clin Ther. 2014;36(7):1104-1111.
doi pubmed - Li K, Hu C, Mei C, Ren Z, Vera JC, Zhuang Z, Jin J, et al. Sequential combination of decitabine and idarubicin synergistically enhances anti-leukemia effect followed by demethylating Wnt pathway inhibitor promoters and downregulating Wnt pathway nuclear target. J Transl Med. 2014;12:167.
doi pubmed - ClinicalTrials.gov. <https://clinicaltrials.gov/ct2/results?recrs=ab&cond=Leukemia&term=decitabine&cntry=&state=&city=&dist=>.
- Dorantes-Acosta E, Pelayo R. Lineage switching in acute leukemias: a consequence of stem cell plasticity? Bone Marrow Res. 2012;2012:406796.
doi pubmed - Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103(5):1635-1640.
doi pubmed - Kopp LM, Ray A, Denman CJ, Senyukov VS, Somanchi SS, Zhu S, Lee DA. Decitabine has a biphasic effect on natural killer cell viability, phenotype, and function under proliferative conditions. Mol Immunol. 2013;54(3-4):296-301.
doi pubmed - Ding K, Fu R, Liu H, Nachnani DA, Shao ZH. Effects of decitabine on megakaryocyte maturation in patients with myelodysplastic syndromes. Oncol Lett. 2016;11(4):2347-2352.
doi pubmed - Li H, Wang L, Wu Y, Su L, Zhao H, Zhang Y, Wang Z, et al. Very-Low-Dose Decitabine Is Effective in Treating Intermediate- or High-Risk Myelodysplastic Syndrome. Acta Haematol. 2017;138(3):168-174.
doi pubmed - Zhao H, Zhu H, Huang J, Zhu Y, Hong M, Zhu H, Zhang J, et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk Res. 2018;66:1-7.
doi pubmed - Valdez BC, Li Y, Murray D, Liu Y, Nieto Y, Champlin RE, Andersson BS. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget. 2018;9(3):3908-3921.
doi pubmed - Sun W, Triche T, Jr., Malvar J, Gaynon P, Sposto R, Yang X, Bittencourt H, et al. A phase 1 study of azacitidine combined with chemotherapy in childhood leukemia: a report from the TACL consortium. Blood. 2018;131(10):1145-1148.
doi pubmed - Ye X, Chen D, Zheng Y, Zhu X, Fu J, Huang J. Effective treatment of low-dose decitabine in myelodysplastic syndrome/myeloproliferative neoplasms. Onco Targets Ther. 2017;10:5425-5428.
doi pubmed - Ai H, Wei XD, Yin QS, Wang P, Mi RH, Yuan FF, Chen L, et al. [The clinical safety and efficacy of low dose subcutaneous decitabine in treating acute myeloid leukemia and intermediate- or higer-risk myelodysplastic syndromes in the elderly patients]. Zhonghua Nei Ke Za Zhi. 2017;56(8):606-609.
- Shi P, Zhang L, Chen K, Jiang Z, Deng M, Zha J, Guo X, et al. Low-dose decitabine enhances chidamide-induced apoptosis in adult acute lymphoblast leukemia, especially for p16-deleted patients through DNA damage. Pharmacogenomics. 2017;18(13):1259-1270.
doi pubmed - Jabbour E, Short NJ, Montalban-Bravo G, Huang X, Bueso-Ramos C, Qiao W, Yang H, et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood. 2017;130(13):1514-1522.
doi pubmed - Bryan J, Kantarjian H, Garcia-Manero G, Jabbour E. Pharmacokinetic evaluation of decitabine for the treatment of leukemia. Expert Opin Drug Metab Toxicol. 2011;7(5):661-672.
doi pubmed - Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102(13):932-941.
doi pubmed - Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496-2506.
doi pubmed - Patnaik MM, Hanson CA, Hodnefield JM, Lasho TL, Finke CM, Knudson RA, Ketterling RP, et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia. 2012;26(1):101-105.
doi pubmed - Yi WR, Li ZH, Qi BW, Ernest ME, Hu X, Yu AX. Downregulation of IDH2 exacerbates the malignant progression of osteosarcoma cells via increased NF-kappaB and MMP-9 activation. Oncol Rep. 2016;35(4):2277-2285.
doi pubmed - Van Damme M, Crompot E, Meuleman N, Maerevoet M, Mineur P, Bron D, Lagneaux L, et al. Characterization of TET and IDH gene expression in chronic lymphocytic leukemia: comparison with normal B cells and prognostic significance. Clin Epigenetics. 2016;8:132.
doi pubmed - Albano F, Anelli L, Zagaria A, Coccaro N, Minervini A, Rossi AR, Specchia G. Decreased TET2 gene expression during chronic myeloid leukemia progression. Leuk Res. 2011;35(11):e220-222.
doi pubmed - Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11-24.
doi pubmed - Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, Masse A, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118(9):2551-2555.
doi pubmed - Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25-38.
doi pubmed - Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289-2301.
doi pubmed - Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32(5):663-669.
doi pubmed - He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. 2011;117(14):3869-3880.
doi pubmed - Wang S, He Q, Ma D, Xue Y, Liu F. Irf4 regulates the choice between T lymphoid-primed progenitor and myeloid lineage fates during embryogenesis. Dev Cell. 2015;34(6):621-631.
doi pubmed - Xu H, Chaudhri VK, Wu Z, Biliouris K, Dienger-Stambaugh K, Rochman Y, Singh H. Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat Immunol. 2015;16(12):1274-1281.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.