

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 6, Number 2-3, September 2017, pages 52-58
Thrombotic Microangiopathy With Granulomatosis Interstitial Nephritis in an Allogenic Bone Marrow Transplant Patient: A Case Report and Review of the Literature
Abu-Sayeef Mirzaa, Sean Vermaa, d, Liying Fub, Claude Bassilc
aDepartment of Internal Medicine, University of South Florida, Tampa, FL, USA
bDepartment of Pathology, Tampa General Hospital, Tampa, FL, USA
cDepartment of Onconephrology, Moffitt Cancer Center, Tampa, FL, USA
dCorresponding Author: Sean Verma, Department of Internal Medicine, University of South Florida, 13220 USF Laurel Drive, Tampa, FL 33612, USA
Manuscript submitted March 29, 2017, accepted April 27, 2017
Short title: TMA in a BMT Patient
doi: https://doi.org/10.14740/jh326e
| Abstract | ▴Top |
Transplant-associated thrombotic microangiopathy (TA-TMA) is a rare complication of hematopoietic stem cell transplantation (HSCT) with variable presentations. TA-TMA has often been described as a diagnosis of exclusion but a renal biopsy is rarely pursued to confirm the diagnosis, an essential step for our patient with renally limited TMA. We report a case report from the onconephrology clinic and review the literature associated with TA-TMA as it relates to diagnosis and treatment. A 45-year-old woman with acute myeloid leukemia and stage 3 chronic kidney disease underwent a matched unrelated donor allogenic HSCT. Postoperatively, she developed gastrointestinal graft versus host disease (GvHD) and was treated with tacrolimus, sirolimus, budesonide, and beclomethasone. Following discharge, she developed uncontrolled hypertension and required losartan, amlodipine, carvedilol, clonidine patch, and hydralazine as needed. On day 180 post-transplant, she developed lower extremity edema and acute kidney injury (AKI) with creatinine increasing to 2 mg/dL. On day 480 post-transplant, she developed worsening thrombocytopenia, anemia, new hematuria, left flank pain, and worsening renal function with creatinine peaking to 6 mg/dL. Peripheral smear revealed no schistocytes, lactate dehydrogenase of 265 mg/dL, and urinalysis with 100 mg/dL protein. ADAMTS 13 activity was normal (92%) and no inhibitor was detected. She became anuric and was started on hemodialysis. Renal biopsy revealed glomerular changes consistent with TA-TMA. During HSCT, systemic vascular endothelial injury triggers microangiopathic hemolytic anemia, platelet consumption, injury of glomerular endothelial cells and fibrin occluded renal capillaries. Thus, TA-TMA should be considered in HSCT patients with elevated LDH, proteinuria, hypertension, and AKI. However, a diagnosis is difficult to confirm without a renal biopsy. Treatment involves discontinuing potentially toxic agents such as calcineurin inhibitors and sirolimus, prescribing adequate antimicrobial treatment, and using renal replacement therapy if needed. A renal biopsy early in the course of disease not only confirms the diagnosis, but may limit the extent of disease.
Keywords: Thrombotic microangiopathy; Bone marrow transplant; Interstitial nephritis; Renal biopsy
| Introduction | ▴Top |
Transplant-associated thrombotic microangiopathy (TA-TMA) is a known, but rare complication of hematopoietic stem cell transplantation (HSCT), occurring in about 10-30% of HSCT patients [1-4]. In a bone marrow transplant (BMT) patient, the cause of TMA can be difficult to discern due to coexisting graft versus host disease (GvHD), transplant complications such as infection, disseminated intravascular coagulation, and side effects from immunosuppression [2]. In addition, TMA can present variably, sometimes without findings of hemolytic anemia or acute kidney injury (AKI) [5]. Few published studies document renally limited TA-TMA. We present a novel case in which progressive renal disease was eventually explained by a renal biopsy which revealed TA-TMA with granulomatous interstitial nephritis more than 1 year after HSCT.
| Case Report | ▴Top |
A 45-year-old woman with an allogeneic stem cell transplant from a matched unrelated donor for chemotherapy-induced high grade myelodysplastic syndrome (MDS) with 5q deletion which subsequently transformed into acute myeloid leukemia (AML) presented to the emergency room with nausea and vomiting. Her dehydration and emesis was treated with intravenous fluids and antiemetics. On admission, her creatinine was 2 mg/dL and her renal function continued to worsen over subsequent days (Fig. 1). About 5 years ago, she was treated for endometrial adenocarcinoma with carboplatin and paclitaxel which was complicated by the development of peripheral neuropathy, and MDS which transformed into AML 3 years later. She underwent induction chemotherapy with cytarabine and high dose daunorubicin (7+3). Due to residual disease on bone marrow biopsy, she was re-induced with salvage chemotherapy consisting of cladribine, cytarabine, and filgrastim (CLAG) with subsequent bone marrow biopsy showing hypocellularity with no blasts. She underwent consolidation chemotherapy with CLAG again and underwent allogenic HSCT.
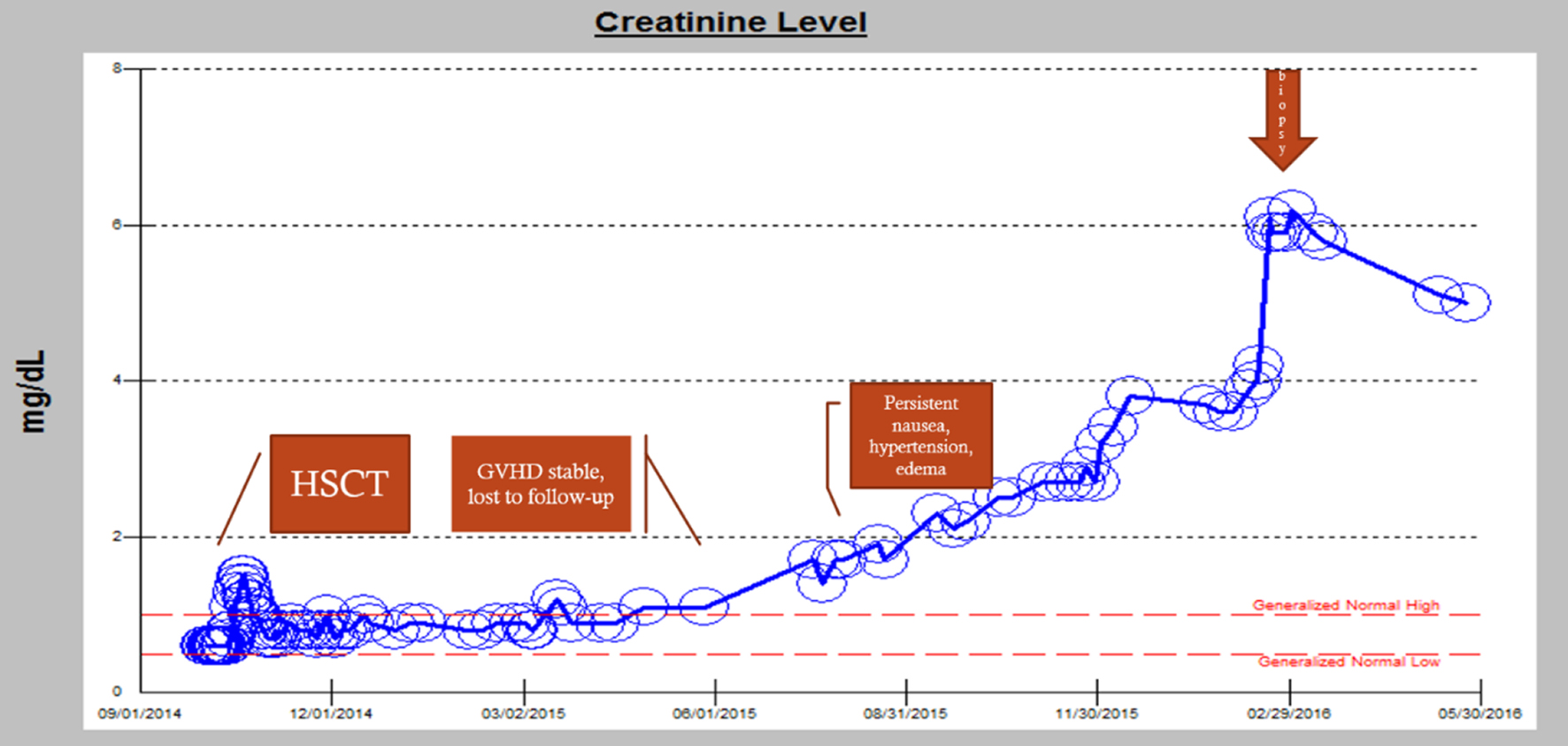 Click for large image | Figure 1. Worsening renal function over time: from transplant to eventual hemodialysis. |
She was given a 5-day course of fludarabine and melphalan for transplant conditioning and tacrolimus with sirolimus for GvHD prophylaxis. She tolerated her HSCT without any side effects. Her hospital course was complicated by presumed acute gastrointestinal GvHD, diagnosed clinically due to worsening nausea, vomiting, and diarrhea and was treated with beclomethasone, budesonide, tacrolimus and sirolimus. She later began having neutropenic fevers, bacteremia treated with vancomycin and also started on amphotericin due to concern for a disseminated fungal infection. During this time, her renal function declined with suspicion regarding vancomycin and amphotericin. At the time of discharge, her creatinine was 1.2 mg/dL.
Approximately 180 days after transplant, her gastrointestinal symptoms resolved and her follow-up visits became inconsistent; however, she continued her GvHD prophylaxis regimen. She presented intermittently with hypertension and peripheral edema which was treated with losartan 100 mg twice daily and furosemide 10 - 20 mg per day as needed. Due to her non-compliance with appointments, she presented to clinic with uncontrolled hypertension and occasional nausea only when she ran out of her immunosuppressants. On subsequent follow-up appointments, her renal function continued to decline and her resistant hypertension required the addition of amlodipine, carvedilol, clonidine patch, and hydralazine.
Approximately 360 days post-BMT, she continued to have uncontrolled hypertension, nausea, vomiting, and weight loss, and developed palpable purpura of her lower extremities. Skin biopsy revealed extravasated erythrocytes consistent with purpura without presence of vasculitis. Tacrolimus was discontinued and she continued on sirolimus and mycophenolate mofetil for immunosuppression. Around 400 days after BMT, she began to experience hypertensive crisis with blood pressure of 198/136 mm Hg and was hospitalized twice due to difficulty tolerating her oral antihypertensives secondary to nausea and emesis. On each admission, she improved with intravenous fluids, antiemetics, and resumed her oral antihypertensives. Losartan was held, but her AKI continued to worsen (Fig. 1).
Around 480 days post-BMT, she developed 2 weeks of hematuria, intermittent lower back pain, and left flank pain. She was seen in nephrology clinic for her chronic kidney disease (CKD). Given her new constellation of anemia, hematuria, flank pain, and persistent proteinuria on urinalysis, there was concern for possible TA-TMA (Fig. 2). An ADAMTS13 revealed activity was normal (92%). Peripheral blood smear was unremarkable without any schistocytes. Her renal function panel showed rapidly progressing renal failure. She became anuric and was started on hemodialysis. Renal biopsy revealed histomorphological features consistent with TMA with granulomatous tubulointerstitial nephritis (Fig. 3). She currently continues to be hemodialysis dependent and follows regularly with nephrology and the BMT clinic.
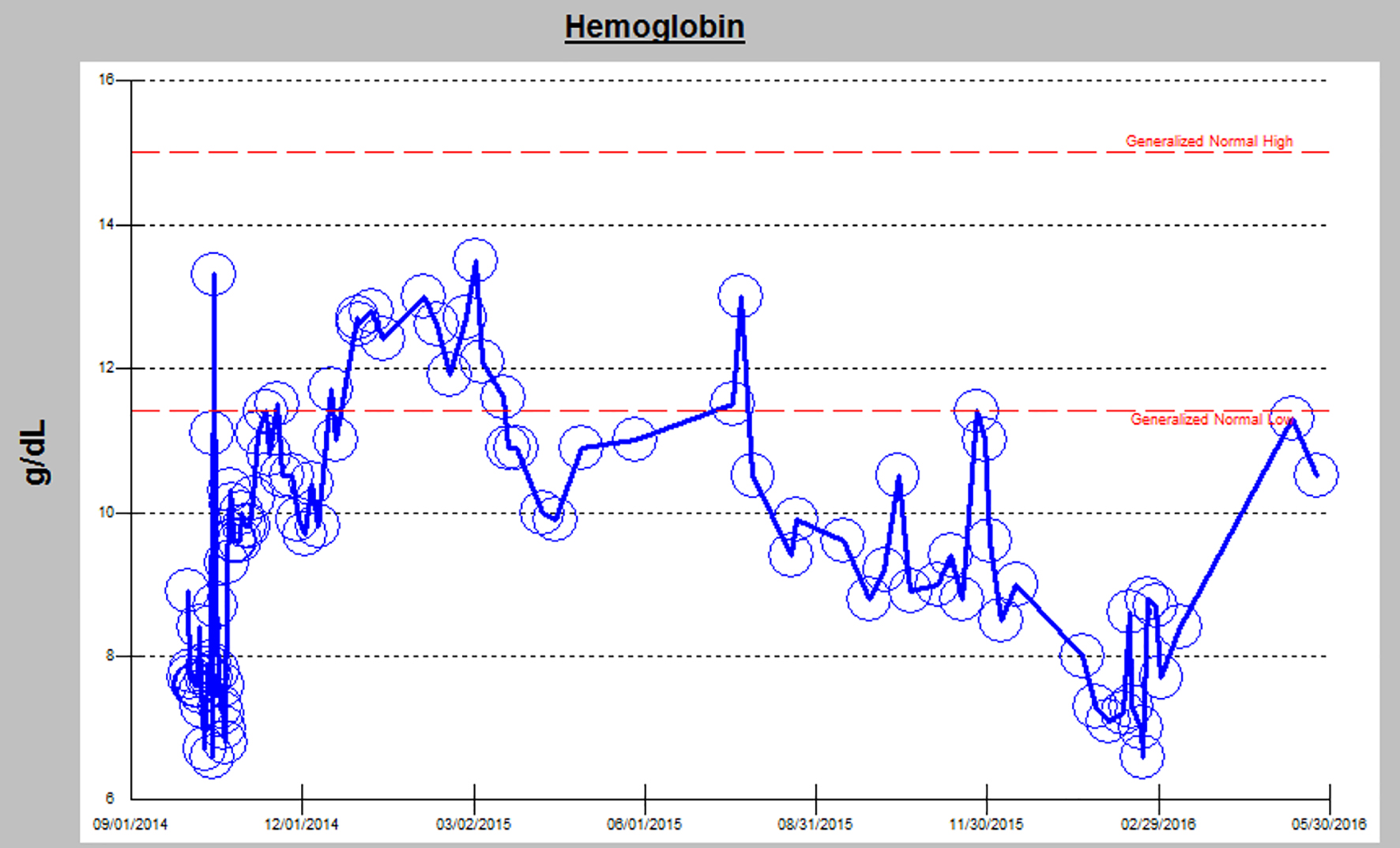 Click for large image | Figure 2. Worsening anemia in correlation with renal decline. |
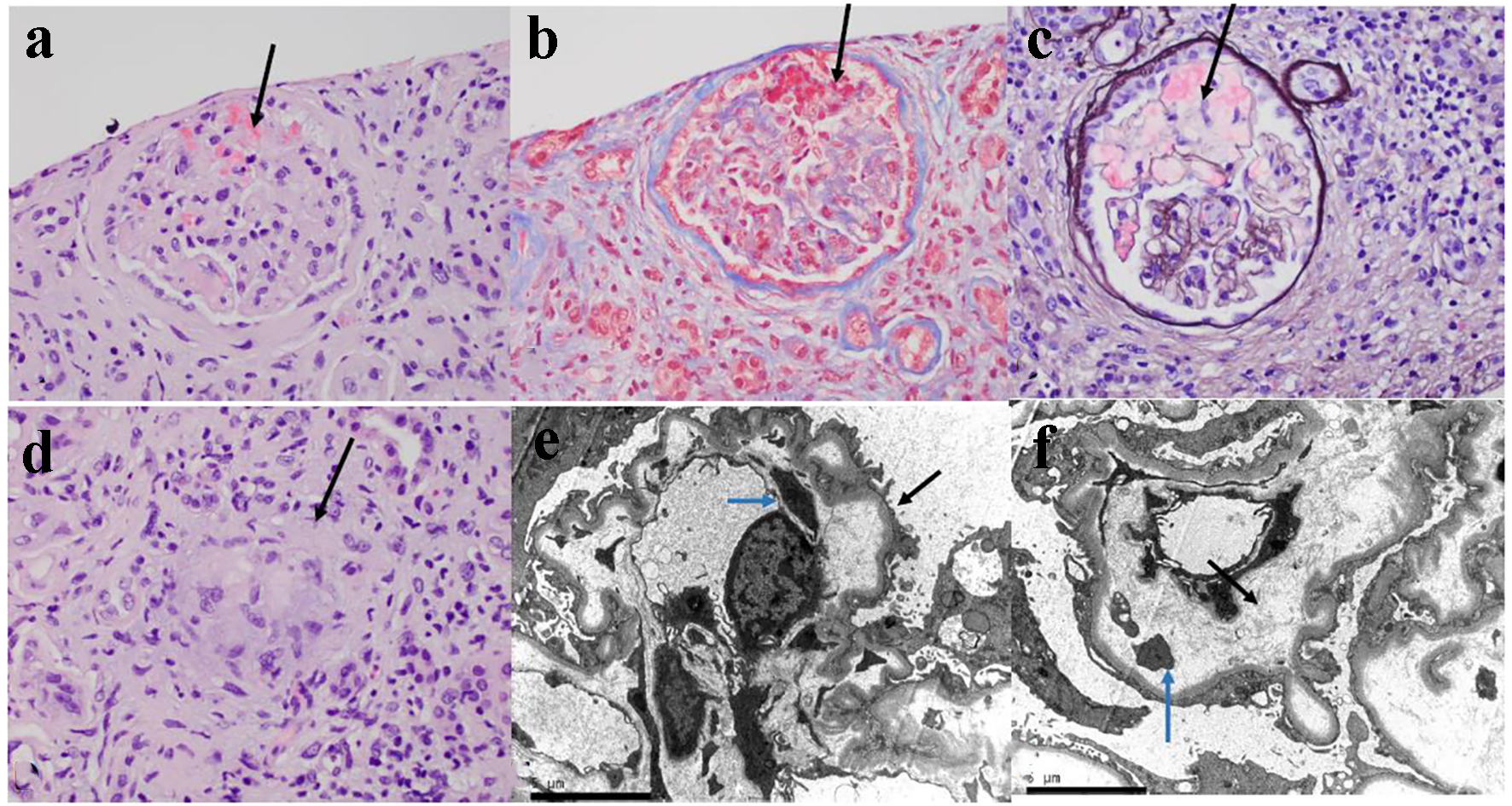 Click for large image | Figure 3. The kidney biopsy tissue of the patient. (a) The glomerulus is slightly hypocellular, and most of the glomerular capillary lumina are closed due to thickening of the capillary walls. Red blood cells and fragmented cells are seen in the mesangial area (H&E, × 400). (b) Trichrome stain showing microthrombi in glomerular capillaries and focal reduplication of the glomerular capillary basement membranes (× 400). (c) Ectatic glomerular capillary lumina are present as a result of mesangiolysis. Focal reduplication of the glomerular capillary basement membranes is also present (Jones silver stain, × 400). (d) Focal non-caseating granuloma is present in the interstitium (black arrow, H&E, × 400). (e) Electron microscopy of glomerular capillary loops showing podocytes vacuolization, extensive effacement of foot processes (black arrow) and basement membrane reduplication (blue arrow). (f) Electron microscopy of glomerular capillary loops showing markedly narrowed capillary lumen and widening of the subendothelial spaces with electrolucent fluffy material (black arrow) and platelets (blue arrow). |
| Discussion | ▴Top |
Overview of TA-TMA
TA-TMA can be caused by a number of factors, making it a difficult etiology to diagnose and treat. It is characterized by systemic vascular endothelial injury that can be triggered by several different elements of the bone marrow transplant process. This endothelial injury causes microangiopathic hemolytic anemia and platelet consumption, which results in thrombosis and fibrin deposition in the microcirculation [6]. TA-TMA is associated with complement system activation; in general, complement dysregulation is associated with poor prognosis. Though common manifestations include hemolytic anemia and thrombocytopenia, organ damage causes the most morbidity and mortality, namely acute renal failure [2]. TA-TMA, in particular, involves injury of glomerular endothelial cells in which fibrin occludes the renal capillaries [4, 7] (Fig. 3).
This endothelial damage in TA-TMA can be caused by a number of factors. It was once assumed that the myeloablative and intense conditioning regimens prescribed prior to HSCT would increase risk of TMA, but this has not been statistically and consistently proven. Infections from aspergillus, cytomegalovirus, parvovirus B19, human herpes virus-6, and even adenovirus, which bind to vascular endothelial growth factor (VEGF) and BK virus, which causes glomerular and renal tubular cell damage, have all been associated with increased incidence of TMA. In addition, coagulation cascade markers and complement activation mediated by the humoral immune response play integral roles in the development of TMA [6].
TA-TMA is difficult to diagnose due to its variation of criteria (the Bone Marrow Transplant Clinical Trials Network versus the International Working Group criteria) [8]. However, it is understood that elevated lactate dehydrogenase, proteinuria on routine urinalysis, and hypertension are the earliest markers of TMA. Proteinuria and evidence of terminal complement activation (elevated sC5b-9) in the blood at the time of TMA diagnosis were associated with very poor survival (20% at 1 year) [5]. AKI usually occurs 1 month after TMA, whereas hypertension and proteinuria typically occurs 10 - 14 days after TMA [5]. Different criteria signify haptoglobin as normal or decreased in TMA, due to the role of haptoglobin in hemolytic anemia as well as its role as an acute phase reactant [2]. Therefore, one should consider TMA in HSCT recipients if they present with an acute elevation in LDH, proteinuria, and hypertension out of proportion to what would be expected from calcineurin inhibitor and steroid therapy [5].
GvHD and TA-TMA
The relationship between GvHD and TA-TMA is known, but not fully understood due to confounding variables. It is thought that during engraftment, donor T lymphocytes cause direct damage to host endothelial cells. Other theories of endothelial injury from transplant include circulating cytokines, low vascular endothelial growth factor (VEGF), and coagulation pathway activation. While the exact pathophysiology of chronic GvHD is unknown, it is often associated with thrombocytopenia [6, 9], which is consistent with our patient (Fig. 4).
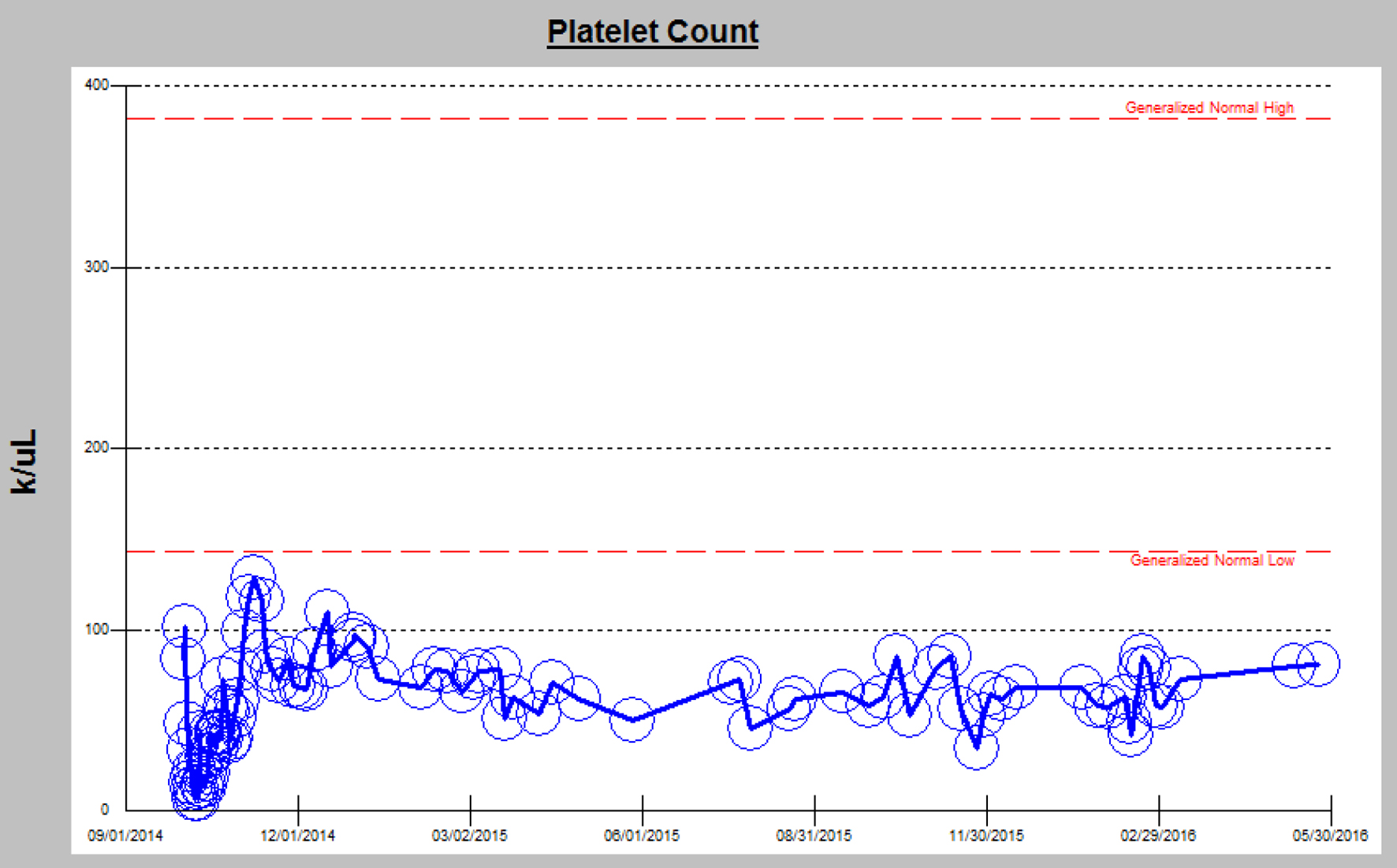 Click for large image | Figure 4. Chronic thrombocytopenia in the setting of chronic GvHD. |
Differentiating between intestinal TMA from acute GvHD can be difficult in patients suffering from severe and refractory diarrhea after BMT due to the similar symptomology. When a patient develops diarrhea after transplantation, it is usually a diagnostic component of GvHD. On the contrary, solitary gut TMA should be in the differential because the treatment differs between TMA and GvHD. GvHD is typically treated with increasing immunosuppression and TMA is treated by decreasing immunosuppression [10]. Although it is understood that the first-line therapy for chronic GvHD is steroids, refractory cases have no treatment guidelines resulting in varying combinations of immunosuppressants often based on expert opinion.
Immunosuppressants: toxicity vs. treatment?
Many drugs are known to cause nephrotoxicity [11]. While renal failure can be due to chemotherapy, radiation, fluid loss secondary to diarrhea or vomiting, sepsis, or nephrotoxicity secondary to calcineurin inhibitors and antimicrobials, there are BMT-specific reasons such as marrow infusion toxicity, hepatic veno-occlusive disease, TMA, and GvHD [12, 13]. Research surrounding drug-induced TMA is still growing as the list of insulting agents vary widely. More research shows that calcineurin inhibitors (cyclosporine and tacrolimus) cause renal toxicity. However, antiangiogenic therapy such as sirolimus targeting VEGF or its receptor has been increasingly associated with TMA [14, 15]. This may be due to the fact that sirolimus is a mTOR inhibitor which is a required component for VEGF production and signaling [16]. Similarly, bevacizumab, a more well-known antiangiogenic agent, causes TMA by binding VEGF [17]. Combining both calcineurin inhibitors and sirolimus results in higher rates of TMA [18].
Though some studies show that using both tacrolimus and sirolimus for GvHD prophylaxis increases risk of TA-TMA, other studies are inconclusive [1]. One autopsy study showed that the association between GvHD and renal TMA was independent of the use of calcineurin inhibitors and conditioning with total-body irradiation [19]. Nevertheless, TMA associated with sirolimus had a more favorable prognosis compared to TMA associated with calcineurin inhibitors alone [18]. Thus, sirolimus has remained the first choice for GvHD control for more than a decade given its superiority in renal outcomes over calcineurin inhibitors and other immunosuppressives such as methotrexate despite its increasing association with TA-TMA [20]. Sirolimus has been associated with greater protection against vasculopathy but at the same time induces proteinuria [16]. This downregulation of VEGF by sirolimus prevents repair of injured renal endothelium and is thought to be a potential predisposing factor for TMA, preferentially affecting the kidneys [21]. The balance between risk and benefit is precarious as providers must decide between using sirolimus for GvHD prophylaxis despite the known and increased risk of TMA.
Renal pathology in GvHD and TA-TMA
Patients who receive HSCT experience extensive damage to their kidneys. Chronic kidney disease is estimated to impact 15-20% HSCT recipients [22]. Acute injury is considered to occur when serum creatinine levels are elevated up to 100 days after transplantation, whereas chronic injury represents injury at or after that [13]. This was the case for our patient who presented with a decline in renal function, more than a year after her transplant. Several theories exist as to why TA-TMA tends to preferentially affect the kidneys. One reason may be that fenestrated endothelium of the glomeruli is vulnerable to damage [23]. Yet an autoimmune process may exist given the fact that inflammatory cells such as CD3+ and CD8+ T cells and cytotoxic T cells with natural killer cells were detected in glomeruli, tubules, and the interstitium of patients with TMA. Endothelial damage in TA-TMA has rarely been reported outside the kidney. Another theory states there is more turbulent blood flow through the renal microvasculature than anywhere else [24].
HSCT recipients often experience a number of concurrent renal abnormalities. According to a recent report, kidney-biopsy specimens obtained from patients with TMA showed mesangiolysis and loss of endothelial cells with occlusion of capillary lumens with fibrin, as was the case for our patient [13]. According to one institution’s study of 67 patients, kidney specimens often demonstrated acute tubular injury, interstitial fibrosis, arteriolar hyaline, arteriosclerosis, and rarely membranous nephropathy [25]. Another study showed that the most commonly reported pathology was membranous nephropathy, followed by minimal change disease [22]. However, our patient presented with a rare entity on renal biopsy: the combination of both TMA and interstitial (granulomatous) nephritis (Fig. 3). Granulomatous interstitial nephritis is detected in less than 1% of all renal biopsies and usually associated with analgesics, antibiotics, and granulomatous disorders [26].
Treatment strategies
Autopsy and biopsy studies show that any clinical criteria for TMA lack sensitivity and specificity for renally limited TMA [22]. The question then becomes, should every BMT patient be getting a renal biopsy? Though it presents similarly to other thrombotic diseases, TA-TMA is treated differently. TA-TMA is different than TTP in that these patients often lack suppression of ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13) and do not respond to plasma exchange [2]. When TA-TMA is suspected, conservative supportive care should be initiated. This includes discontinuing potentially toxic agents such as calcineurin inhibitors and sirolimus, prescribing adequate antimicrobial treatment, and maintaining adequate renal function using renal replacement therapy [4]. Whereas some clinicians discontinue calcineurin inhibitors and opt for switching to mycophenolate mofetil, sirolimus or both, some patients have experienced resolution of their TMA when given increasing doses of calcineurin inhibitors [13]. Furthermore, aggressively managing hypertension, another risk factor for CKD and TA-TMA, is imperative as 70% of children and adults develop hypertension within the first 2 years of their HSCT [13]. Though some studies show blocking the complement system with eculizumab is a possible treatment for TA-TMA, the evidence is still limited as to whether it improves outcomes in patients [27].
Conclusions
This case study emphasizes the importance of TA-TMA as a consideration in the differential diagnosis in patients with HSCT who develop hematuria, proteinuria, hypertension (especially hypertension that is resistant), AKI, and elevated LDH, or a combination of these signs. This case also underscores the importance of renal biopsy when diagnosing the etiology of declining renal function status post HSCT. There is a definite need for biopsy in BMT patients with renal dysfunction. Though biopsy results can reveal type, acuity, and chronicity of the renal disease which facilitate treatment decisions, cancer patients are not often biopsied. TA-TMA is a complex and difficult diagnosis that requires a high degree of suspicion from both the hematologist and nephrologist when managing the post-HSCT patient.
Acknowledgments
Special thanks and appreciation are given to the Pathology Department of the Tampa General Hospital for supplying the histopathology for this patient. Also, we appreciate the Onconephrology Clinic at Moffitt Cancer Center where this patient was first clinically diagnosed with TA-TMA and continues to be routinely followed and managed.
Disclosures
None.
Funding
None.
| References | ▴Top |
- Labrador J, Lopez-Godino O, Lopez-Corral L, et al. Severe Gut Acute Graft Versus host Disease (GVHD) Influence Thrombotic Microangiopathy in 86 Allogeneic Hematopoietic Stem Cell Recipients Who Received Tacrolimus-Based Regimen for GVHD Prophylaxis. Blood. 2015;120(21):1955.
- Chapin J, Shore T, Forsberg P, Desman G, Van Besien K, Laurence J. Hematopoietic transplant-associated thrombotic microangiopathy: case report and review of diagnosis and treatments. Clin Adv Hematol Oncol. 2014;12(9):565-573.
pubmed - Schwarz A, Haller H, Schmitt R, Schiffer M, Koenecke C, Strassburg C, Lehner F, et al. Biopsy-diagnosed renal disease in patients after transplantation of other organs and tissues. Am J Transplant. 2010;10(9):2017-2025.
doi pubmed - Rosenthal J. Hematopoietic cell transplantation-associated thrombotic microangiopathy: a review of pathophysiology, diagnosis, and treatment. J Blood Med. 2016;7:181-186.
doi pubmed - Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J, Myers K, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood. 2014;124(4):645-653.
doi pubmed - Laskin BL, Goebel J, Davies SM, Jodele S. Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood. 2011;118(6):1452-1462.
doi pubmed - Haddad G, Zhabyeyev P, Farhan M, Zhu LF, Kassiri Z, Rayner DC, Vanhaesebroeck B, et al. Phosphoinositide 3-kinase beta mediates microvascular endothelial repair of thrombotic microangiopathy. Blood. 2014;124(13):2142-2149.
doi pubmed - Zheng W, Luo Y, Tan Y, et al. Evaluation the Risk Factor of Transplantation-Associated Thrombotic Microangiopathy. Blood. 2015;116(21):4527.
- Hill W, Sotlar K, Kolb HJ, Hausmann A, Tischer J, Rank A. Does Microangiopathy Predict Chronic GvHD After Allogeneic Stem Cell Transplantation? A Clinical and Immunohistochemical Study on Bone Marrow Biopsies. Blood. 2015;118(21):4540.
- Miyamura K, Yamada M, Nishida T, et al. Clinical and Pathological Diagnosis of Transplantation Associated Microangiopathy of Gut: Successful Reduction of Immunosuppressant Which Is Opposite Treatment of GVHD. Blood. 2015;108(11):2972.
- Ghane Shahrbaf F, Assadi F. Drug-induced renal disorders. J Renal Inj Prev. 2015;4(3):57-60.
pubmed - Kemmner S, Verbeek M, Heemann U. Renal dysfunction following bone marrow transplantation. J Nephrol. 2017;30(2):201-209.
doi pubmed - Hingorani S. Renal Complications of Hematopoietic-Cell Transplantation. N Engl J Med. 2016;374(23):2256-2267.
doi pubmed - den Deurwaarder ES, Desar IM, Steenbergen EJ, Mulders PF, Wetzels JF, van Herpen CM. Kidney injury during VEGF inhibitor therapy. Neth J Med. 2012;70(6):267-271.
pubmed - Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8(2):128-135.
doi pubmed - Ko HT, Yin JL, Wyburn K, Wu H, Eris JM, Hambly BD, Chadban SJ. Sirolimus reduces vasculopathy but exacerbates proteinuria in association with inhibition of VEGF and VEGFR in a rat kidney model of chronic allograft dysfunction. Nephrol Dial Transplant. 2013;28(2):327-336.
doi pubmed - Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129-1136.
doi pubmed - Cutler C, Henry NL, Magee C, Li S, Kim HT, Alyea E, Ho V, et al. Sirolimus and thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11(7):551-557.
doi pubmed - Changsirikulchai S, Myerson D, Guthrie KA, McDonald GB, Alpers CE, Hingorani SR. Renal thrombotic microangiopathy after hematopoietic cell transplant: role of GVHD in pathogenesis. Clin J Am Soc Nephrol. 2009;4(2):345-353.
doi pubmed - Cutler C, Antin JH. Sirolimus for GVHD prophylaxis in allogeneic stem cell transplantation. Bone Marrow Transplant. 2004;34(6):471-476.
doi pubmed - Sartelet H, Toupance O, Lorenzato M, Fadel F, Noel LH, Lagonotte E, Birembaut P, et al. Sirolimus-induced thrombotic microangiopathy is associated with decreased expression of vascular endothelial growth factor in kidneys. Am J Transplant. 2005;5(10):2441-2447.
doi pubmed - Troxell ML, Higgins JP, Kambham N. Renal pathology associated with hematopoietic stem cell transplantation. Adv Anat Pathol. 2014;21(5):330-340.
doi pubmed - Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676-1687.
doi pubmed - Fang CJ, Richards A, Liszewski MK, Kavanagh D, Atkinson JP. Advances in understanding of pathogenesis of aHUS and HELLP. Br J Haematol. 2008;143(3):336-348.
doi pubmed - Brinkerhoff BT, Houghton DC, Troxell ML. Renal pathology in hematopoietic cell transplant recipients: a contemporary biopsy, nephrectomy, and autopsy series. Mod Pathol. 2016;29(6):637-652.
doi pubmed - Shah S, Carter-Monroe N, Atta MG. Granulomatous interstitial nephritis. Clin Kidney J. 2015;8(5):516-523.
doi pubmed - Fernandez C, Lario A, Fores R, Cabrera R. Eculizumab Treatment in a Patient with Hematopoietic Stem Cell Transplantation-Associated Thrombotic Microangiopathy and Steroid-Refractory Acute Graft Versus Host Disease. Hematol Rep. 2015;7(4):6107.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.