

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 4, Number 3, September 2015, pages 214-218
A Rare Variant of Aggressive T-Cell Large Granular Lymphocyte Leukemia Associated With Hepatic Fibrosis and Trisomy 8: A Case Report and Literature Review
Sanjay de Mela, e, Benjamin Wongb, Leena Golec, Siok Bian Ngb, d, Evelyn Koayb, c, Christopher Ng Wai Siongb, Ju Ee Seetb, Aileen Weeb, Wee Joo Chnga, d, Lip Kun Tana, c
aDepartment of Haematology-Oncology, National University Cancer Institute, National University Health System, Singapore
bDepartment of Pathology, National University Health System, Singapore
cDepartment of Laboratory Medicine, National University Health System, Singapore
dCancer Science Institute, Singapore
eCorresponding Author: Sanjay de Mel, Department of Haematology-Oncology, National University Cancer Institute, National University Health System, Singapore
Manuscript accepted for publication August 18, 2015
Short title: Rare Variant of T-LGL
doi: http://dx.doi.org/10.14740/jh223w
| Abstract | ▴Top |
Aggressive T-cell large granular lymphocytic leukemia (T-LGL) is a rare entity with a poor prognosis, the main clinical findings being splenomegaly, lymphadenopathy, thrombocytopenia and a CD3+/CD56+ phenotype. A 21-year-old lady presented with hepatosplenomegaly, lymphadenopathy, “B” symptoms and thrombocytopenia. She subsequently developed jaundice, ascites and deranged liver function. LGL was detected in the peripheral blood and marrow. They had an aberrant CD3+/CD4-/CD8dim/CD56+ phenotype. Karyotyping showed trisomy 8 which has not previously been reported in aggressive T-LGL. A liver biopsy showed hepatic fibrosis with a sinusoidal LGL infiltrate. Her ascitic fluid also showed involvement by T-LGL. She had a poor response to cyclophosphamide, doxorubicin, vincristine and prednisolone (CHOP) chemotherapy and opted for palliation. We performed targeted sequencing to detect mutations in the STAT3, STAT5b and PTPRT genes which were absent, suggesting the presence of an alternative genetic driver in our patient. We report an unusual case of aggressive T-LGL with an aberrant immunophenotype, trisomy 8 and hepatic fibrosis with absence of mutations in the STAT pathway. Our case describes novel features of aggressive T-LGL and the need for further studies to elucidate the genetic basis of this rare disease.
Keywords: Large granular lymphocyte; Leukemia; Hepatic perisinusoidal fibrosis; CD56; Trisomy 8
| Introduction | ▴Top |
T-cell large granular lymphocytic leukemia (T-LGL) is a rare lymphoproliferative disorder characterized by persistent, monoclonal large granular lymphocytosis with a characteristic immunophenotype [1]. The association of T-LGL with autoimmune disorders suggests that sustained immune stimulation and dysregulated FAS ligand-mediated apoptosis may be important in the pathogenesis [2]. Symptoms are usually due to anemia or neutropenia and autoimmune manifestations such as rheumatoid arthritis are common [2]. Splenomegaly is the most common finding on physical examination [2]. The majority of patients have an indolent clinical course, although rare cases of aggressive disease have been described [3]. The diagnosis is established by the presence of circulating large granular lymphocytes (LGL) above 0.4 × 109/L with an activated T cell immunophenotype (CD3+/CD8+/CD56-/CD57+ and/or CD16+) [3]. Clonality can be demonstrated by T-cell receptor (TCR) gene rearrangement studies while bone marrow (BM) studies may be required in patients with low circulating LGL numbers in the right clinical context [3]. Therapeutic options for indolent disease include immunosuppressive therapy with responses reported using methotrexate, cyclosporine and cyclophosphamide [3].
Although hepatic involvement in T-LGL has been reported, association with liver fibrosis has not to our knowledge been previously described [4]. We report a case of aggressive T-LGL associated with hepatic fibrosis and trisomy 8 on karyotyping with an atypical immunophenotype.
| Case Report | ▴Top |
A previously well 21-year-old Vietnamese lady presented in May 2012 with bruising, menorrhagia and abdominal distension of 5 months duration. She also reported fevers of 39 °C associated with weight loss of 9 kg and night sweats. Physical examination revealed hepatosplenomegaly with ascites. Initial investigations in Vietnam revealed a hemoglobin of 9 g/dL, a white cell count of 4.0 × 109/L and a platelet count of 34 × 109/L. Serology tests for hepatitis B, C, HIV, cytomegalovirus and Epstein-Barr virus were negative. Anti-nuclear antibody and anti-double stranded DNA were negative, lactate dehydrogenase was normal and a direct Coomb’s test was negative. Bone marrow aspiration (BMA) was not conclusive.
As her blood counts remained stable, she was managed expectantly until June 2012 when she underwent a splenectomy in view of persistent splenomegaly and worsening thrombocytopenia. Histopathology of the spleen revealed “lymphoid proliferation” but no evidence of malignancy. Her fever and thrombocytopenia improved transiently after the splenectomy but recurred in August 2012 at which point she was treated with pulsed methylprednisolone for 1 week with a transient improvement in the cytopenias. In October 2012, she was found to have worsening jaundice with conjugated hyperbilirubinemia. A fibroscan score of F4 was recorded and she underwent a liver biopsy which suggested cholestatic liver injury with cirrhosis.
She was transferred to our center in January 2013 for further evaluation of her thrombocytopenia. At the time her main symptoms were those of worsening jaundice, ascites, fever and night sweats. She also described menorrhagia and gingival bleeding. Physical examination revealed marked cachexia with ascites, hepatomegaly and scleral icterus. She also had inguinal lymphadenopathy.
Her investigations were repeated, a full blood count showed hemoglobin 11.8 g/dL, white cell count 14.5 × 109/L, neutrophils 5.3 × 109/L, lymphocytes 5.0 × 109/L and platelets 57 × 109/L. Liver function tests showed serum albumin of 34 g/L (38 - 48 g/L), elevated bilirubin at 40 µmol/L (5 - 30 µmol/L) and predominantly conjugated (32 µmol/L). Aspartate transaminase and alanine transaminase were within normal limits. She was coagulopathic with a prothrombin time of 16.8 s (12.0 - 14.5 s) and an activated partial thromboplastin time of 42 s (27.0 - 35.6 s).
The peripheral blood film revealed target cells and a large granular lymphocytosis (Fig. 1A). Her BMA showed LGL comprising 9% of nucleated cells (Fig. 1B). The BM trephine showed a sinusoidal infiltrate of small to medium sized lymphocytes with irregular nuclei (Fig. 1C). Flow cytometry of the BM showed an abnormal population of large T cells with the following phenotype: CD3+/CD4-/CD5-/CD8dim/CD56+/cyGranzymeB+/cyPerforin-/CD16+. The aberrant CD8dim/CD5-/cyPerforin- phenotype supported the suspicion of a neoplastic T-LGL population (Fig. 2A). A 20-metaphase cytogenetic analysis of the BM revealed a karyotype of 46,XX,+8,-11,-20,+mar(cp3)/46,XX(17), indicating three metaphases with trisomy 8, loss of chromosomes 11 and 20, along with a marker chromosome (Fig. 2B). TCR gene rearrangement studies from the BM sample confirmed the presence of a clonal T-cell population. In view of the presence of trisomy 8 on karyotyping, we performed immunohistochemical staining for C-Myc on the patient’s BM trephine, and the malignant cells did not stain positive for C-Myc.
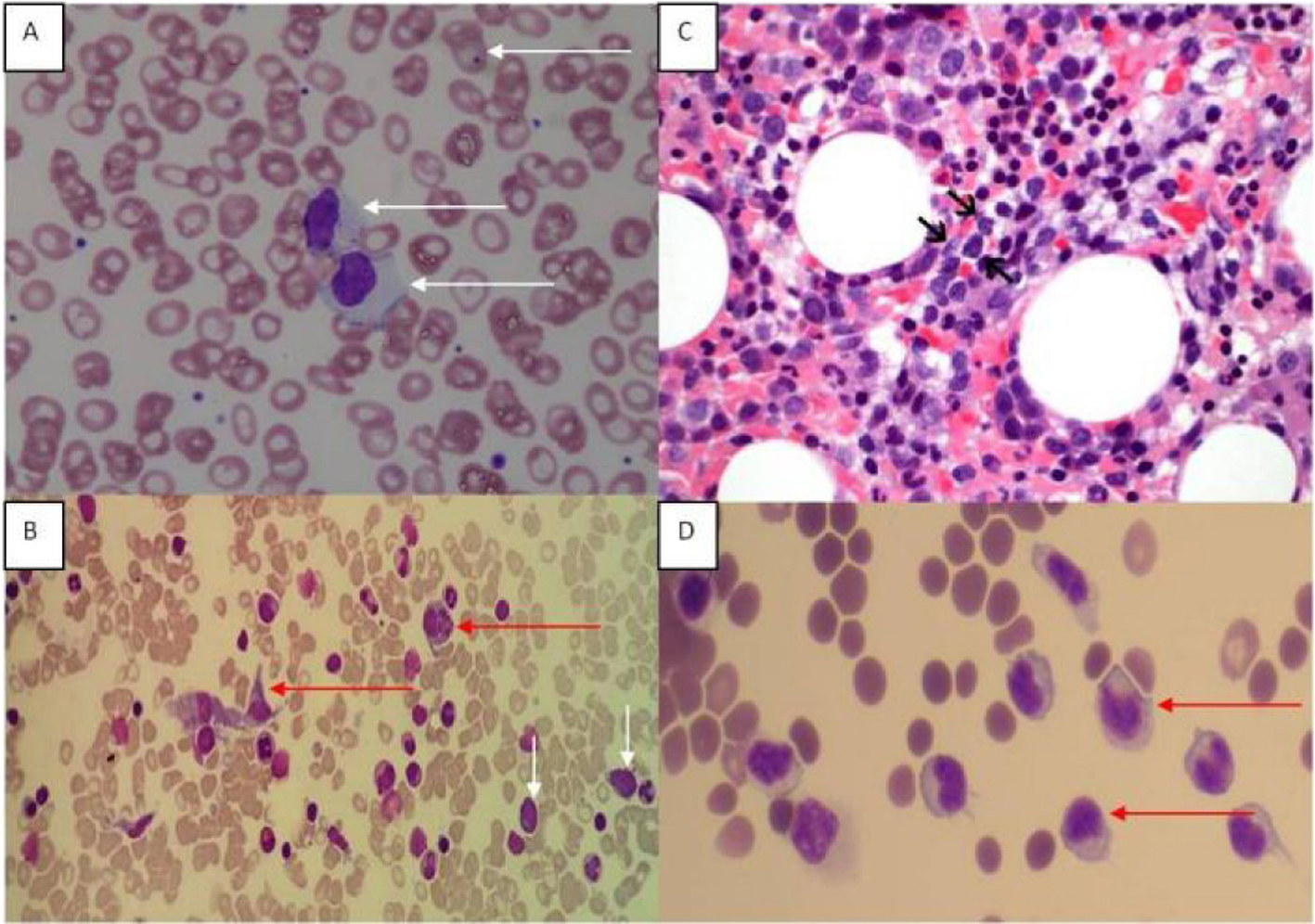 Click for large image | Figure 1. (A) Peripheral blood film showing large granular lymphoid cells. Howell Jolly bodies are seen in some red cells in keeping with a post-splenectomy picture. (B) Bone marrow aspirate (MGG stain) showing hemophagocytic activity and atypical lymphoid cells. Red arrows point to histiocytes, and white arrows indicate atypical lymphoid cells. (C) Bone marrow trephine showed a sinusoidal infiltrate of small to medium sized lymphocytes (arrows) with irregular nuclei and indistinct nucleoli (H&E, original magnification, × 600). (D) Ascitic fluid showing atypical lymphoid cells. |
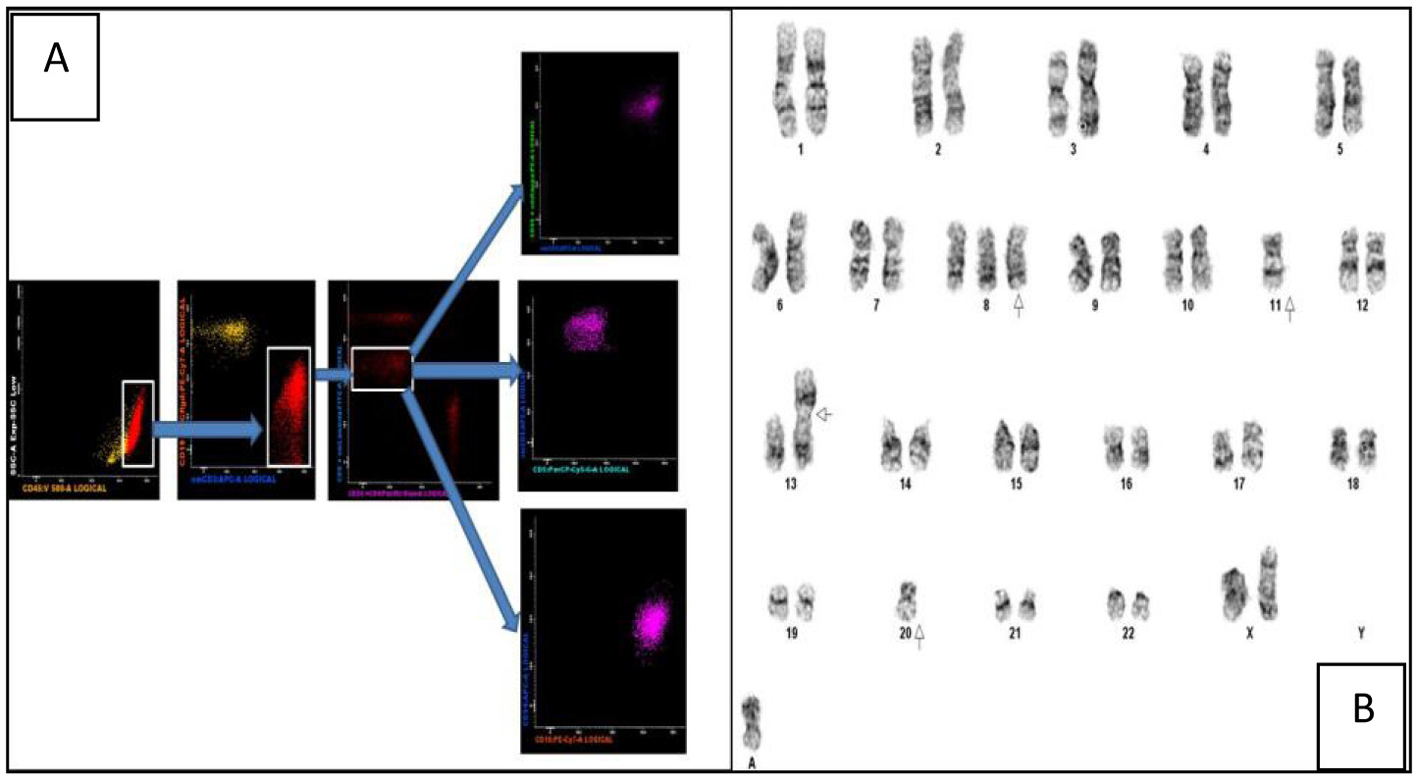 Click for large image | Figure 2. (A) Multiparametric flow cytometric analysis of the bone marrow specimen. The abnormal events are CD45+ with a low side scatter. They are T cells as evidenced by CD3 positivity and lack of CD19 expression (the yellow events in the CD19/CD3 scatter plot are normal B cells). Among the CD8+ T cells there is an abnormal CD8dim sub-population which constitutes the neoplastic clone. These events are CD5-/CD56+/CD16+ and CD94-. (B) Karyogram showing trisomy 8, loss of chromosomes 11 and 20, a non-clonal abnormality of chromosome 13 and a marker chromosome. |
A CT scan of the abdomen showed an enlarged caudate lobe of the liver and extensive ascites with inguinal lymphadenopathy. A liver biopsy was repeated and showed diffuse perisinusoidal fibrosis, extramedullary hematopoiesis and CD3+ lymphocytic infiltrate (Fig. 3). These were morphologically larger than normal lymphocytes and were negative for CD4 and CD8 on immunohistochemistry, strongly suggesting that these were T-LGL infiltrating the sinusoids.
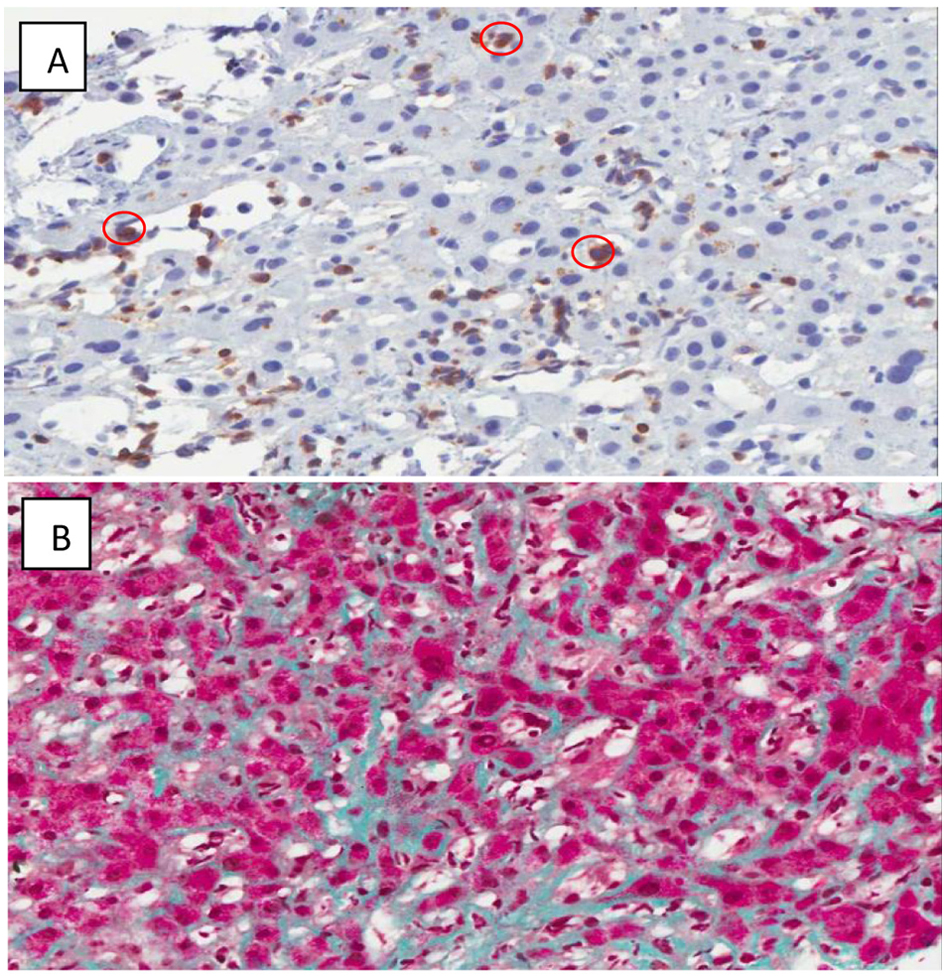 Click for large image | Figure 3. (A) CD 3 Immunoperoxidase highlights the T-cells circled in red. (× 400) Some T-cells are somewhat larger than others. (B) Sinusoidal fibrosis. (Masson's trichrome stain, × 200) The hepatocytes are red, and the collagen is green. Collagen is normally restricted to portal tracts and the cuff of central veins, but here is seen running along sinusoids between plates of hepatocytes. |
She was treated with six courses of cyclophosphamide, doxorubicin, vincristine and prednisolone (CHOP) chemotherapy (she was not fit enough to receive a more aggressive treatment protocol) and had a transient response with an improvement of her cytopenias. Within 6 months of completing her treatment, she developed a recurrence of cytopenias, T-LGL lymphocytosis and worsening ascites and lymphadenopathy. Cytology of the ascitic fluid showed involvement by T-LGL (Fig. 1D); the flow cytometric immunophenotype of the lymphoid cells detected in the ascitic fluid was identical to that seen in the BM previously.
In view of the availability of a clinical trial of a STAT3 inhibitor in our center, we performed targeted sequencing of the patient’s diagnostic BM sample to identify STAT3 mutations which would have made her a candidate for the clinical trial. Unfortunately the STAT3 mutation was negative which made her ineligible for the study. She subsequently returned to Vietnam for palliative treatment.
| Discussion | ▴Top |
Most patients with T-LGL have an indolent clinical course; although aggressive T-LGL has been reported, these cases are rare [5]. Gentile et al reported three patients with aggressive LGL leukemia - the main clinical features present in all three cases were splenomegaly, lymphadenopathy and B symptoms [6]. Recurrent infections and rheumatoid arthritis which are characteristic of indolent LGL were notably absent in these patients [6]. Neutropenia which is commonly reported in indolent T-LGL was not reported in any of the patients while all three had thrombocytopenia which is unusual in indolent T-LGL [6]. Peripheral blood immunophenotyping revealed a CD3+/CD4-/CD8-/CD16+/CD57-/CD56+ expression in this series [6]. Based on the differences in clinical features and consistent CD3+/CD56+ phenotype, the authors proposed CD3+/56+ aggressive T-LGL as a distinctive clinical entity [6]. Matutes and colleagues described a case of T-LGL which transformed into a large T-cell lymphoma 11 years from the original diagnosis [7]. The main clinical features at transformation were those of splenomegaly, fever and lymphadenopathy with a CD3+/CD4-/CD8+/CD56+ T-cell phenotype on immunohistochemistry of the lymph node biopsy [7]. Our patient had clinical features similar to the cases described by Gentile et al as well as a CD3+/CD56+ phenotype. Given the CD4-/8dim phenotype, hepatic infiltration and aggressive disease course, hepatosplenic T-cell lymphoma (HTL) was considered a differential diagnosis. The T cells were however negative for TCR-γ/δ which together with the positive expression of granzyme B (rare in HTL) makes T-LGL the more likely diagnosis [1].
Cytogenetic abnormalities associated with T-LGL have not been clearly defined, mostly due to the rarity of the disease and the classification of some cases as T-chronic lymphocytic leukemia [8]. Loughran et al described two cases of indolent T-LGL with trisomy 14 and trisomy 8, respectively [9]. Trisomy 8 was also described in a case of CD3- NK- LGL leukemia with aggressive transformation [10]. Trisomy 8 has been shown to predict an adverse outcome in acute myeloid leukemia and has been implicated in blast phase transformation of chronic myeloid leukemia [11, 12]. Amplification of the C-Myc oncogene located on chromosome 8q has been postulated as a mechanism of the adverse outcome associated with this cytogenetic abnormality [12]. Ours is the second report of trisomy 8 in T-LGL and the first in the rare, aggressive subset of this disease. It is possible that trisomy 8 may play a role in the transformation of T-LGL from an indolent to an aggressive disease and may have prognostic significance.
Hepatic fibrosis has not previously been described in T-LGL. Loughran et al described four patients with aggressive T-LGL, two of whom underwent a liver biopsy in view of abnormal liver function tests [13]. A neoplastic lymphoid infiltrate was detected in the hepatic sinusoids, with hepatocyte apoptosis demonstrated by TUNEL positivity [13]. It was postulated that the hepatocyte apoptosis was mediated by dysregulated FAS-FAS ligand signaling, as this was supported by the finding of increased serum FAS ligand levels and FAS being expressed on the hepatocytes [13]. They also demonstrated hepatocyte and pneumocyte apoptosis in FAS ligand transgenic mice [13]. The reason for hepatic fibrosis in our patient was not clear. It is possible that the fibrosis was a response to hepatocyte injury resulting from T-LGL infiltration. As dysregulated expression of growth factors such as transforming growth factor β has been shown to be important in the pathogenesis of hepatic fibrosis, it may be postulated that T-LGL infiltration leads to aberrant expression of such mediators [14].
Mutations in the STAT3 (signal transducer and activator of transcription 3) gene have been demonstrated in LGL [15]. Whole exome sequencing (WES) of 77 patients with LGL identified STAT3 mutations in 31 patients (40%), Y640F in 22 (17%), D661Y in 12 (9%) and N647I in 12 (9%). The presence of STAT3 mutations was associated with neutropenia and rheumatoid arthritis in this cohort. In another study looking at T and NK LGL patients, it was shown that 48 of 170 (28%) of patients had a STAT3 mutation, with similar incidence among T and NK subtypes of LGL [16]. Importantly it was demonstrated that even patients without STAT3 mutations had increased expression of STAT3 target genes [15]. The gene expression profile of LGL patients with and without STAT3 mutations had also been shown to be similar [17]. This led to the hypothesis that other genes involved in STAT3 signaling may be dysregulated in patients with LGL without STAT3 mutations. Protein tyrosine phosphatase receptor 3 (PTPR3) is a tumor suppressor gene whose normal function is to reverse tyrosine phosphorylation on STAT3 leading to a deactivation of STAT3 signaling. WES of a patient with LGL without STAT3 mutations identified a missense mutation of PTPR3 (V995M). It was hypothesized that this PTPR3 mutation may be responsible for abnormal STAT3 signaling in LGL patients who lack STAT3 mutations [17].
Rajala and colleagues recently demonstrated that STAT5b mutations may be a driving genetic lesion in LGL without STAT3 mutations [18]. They described three T-LGL patients with STAT5b mutations, two of them had the Y665F mutation while the other one had the N642H mutation. The patients with the Y665F mutation had an indolent clinical course with no treatment required but the N642H patient had a very aggressive disease with extensive organ infiltration and a poor response to chemotherapy. Interestingly the N642H patient had a CD3+, CD56+ phenotype similar to our patient [18].
Given the clinical and immunophenotypic similarity of our patient with the STAT5b N642H mutated patient described above, we performed targeted capillary sequencing to detect somatic missense mutations in the PRPRT and STAT5b genes. Primer sequences were adapted from previous reports [17, 18], but PCR conditions for targeted capillary sequencing of STAT5b exons 14, 15, 16, 17, 18 and PTPRT exon 23, were optimized in-house. We did not identify any mutations at these loci (and she had no STAT3 mutations) suggesting that an alternative genetic driver may be present in our patient.
Although STAT3 inhibitors are in clinical trials, studies investigating STAT5b inhibition are awaited. It is important to further clarify the underlying genetic abnormalities in STAT3 mutation negative LGL, especially those with the rare aggressive variant like ours who had a suboptimal response to conventional chemotherapy [19].
Conclusion
In conclusion, our patient has a rare aggressive variant of T-LGL with the unusual features of trisomy 8 on cytogenetics and hepatic fibrosis. This case adds to the limited data on aggressive T-LGL and supports the proposal that CD3+/CD56+ aggressive T-LGL is a distinct clinical entity. Further research is required to better define the clinicopathological and genetic features of aggressive T-LGL and elucidate the effects of tissue infiltration by this rare leukemia. There is an urgent need for international clinical trials to determine the optimal therapy for this aggressive disease. For young patients, early consideration of stem cell transplant is important in view of the poor outcome with currently available therapy.
Acknowledgement
The authors would like to acknowledge Dr. Kieron Lim, Department of Medicine, and Dr. Liu Te Chih, Department of Laboratory Medicine, National University Health System, Singapore for their assistance in the preparation of the case report.
Conflict of Interest
None.
Financial Support
None.
| References | ▴Top |
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. (Eds.): WHO Classification of Tumours of Haematopoeitic and Lymphoid Tissues. IARC Lyon. 2008.
- Zhang D, Loughran P. Large Granular Lymphocytic Leukaemia, Molecular Pathogenesis, Clinical Manifestations and Treatment. ASH Education Program Book. 2012.
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist. 2004;9(3):247-258.
doi - Loughran TP, Jr. Clonal diseases of large granular lymphocytes. Blood. 1993;82(1):1-14.
pubmed - Lamy T, Loughran TP. Large Granular Lymphocyte Leukemia. Cancer Control. 1998;5(1):25-33.
- Gentile TC, Uner AH, Hutchison RE, Wright J, Ben-Ezra J, Russell EC, Loughran TP, Jr. CD3+, CD56+ aggressive variant of large granular lymphocyte leukemia. Blood. 1994;84(7):2315-2321.
pubmed - Matutes E, Wotherspoon AC, Parker NE, Osuji N, Isaacson PG, Catovsky D. Transformation of T-cell large granular lymphocyte leukaemia into a high-grade large T-cell lymphoma. Br J Haematol. 2001;115(4):801-806.
doi pubmed - Wong KF, Chan JC, Liu HS, Man C, Kwong YL. Chromosomal abnormalities in T-cell large granular lymphocyte leukaemia: report of two cases and review of the literature. Br J Haematol. 2002;116(3):598-600.
doi pubmed - Loughran TP, Jr., Kadin ME, Starkebaum G, Abkowitz JL, Clark EA, Disteche C, Lum LG, et al. Leukemia of large granular lymphocytes: association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann Intern Med. 1985;102(2):169-175.
doi pubmed - Ohno Y, Amakawa R, Fukuhara S, Huang CR, Kamesaki H, Amano H, Imanaka T, et al. Acute transformation of chronic large granular lymphocyte leukemia associated with additional chromosome abnormality. Cancer. 1989;64(1):63-67.
doi - Wolman SR, Gundacker H, Appelbaum FR, Slovak ML. Impact of trisomy 8 (+8) on clinical presentation, treatment response, and survival in acute myeloid leukemia: a Southwest Oncology Group study. Blood. 2002;100(1):29-35.
doi pubmed - Jennings BA, Mills KI. c-myc locus amplification and the acquisition of trisomy 8 in the evolution of chronic myeloid leukaemia. Leuk Res. 1998;22(10):899-903.
doi - Lamy T, Bauer FA, Liu JH, Li YX, Pillemer E, Shahidi H, Gregory SA, et al. Clinicopathological features of aggressive large granular lymphocyte leukaemia resemble Fas ligand transgenic mice. Br J Haematol. 2000;108(4):717-723.
doi pubmed - Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10(1):76-99.
doi - Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, Lagstrom S, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913.
doi pubmed - Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, Olson T, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120(15):3048-3057.
doi pubmed - Andersson EI, Rajala HL, Eldfors S, Ellonen P, Olson T, Jerez A, Clemente MJ, et al. Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT-pathway and T-cell activation. Blood Cancer J. 2013;3:e168.
doi pubmed - Rajala HL, Eldfors S, Kuusanmaki H, van Adrichem AJ, Olson T, Lagstrom S, Andersson EI, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541-4550.
doi pubmed - Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, Li Y, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest. 2001;107(3):351-362.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.