

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 4, Number 1, March 2015, pages 148-150
Acquired β-Thalassemia as an Etiology of Microcytic Anemia in Primary Myelofibrosis
Maya Kanasakia, Takakazu Higuchib, c, Keisuke Kawamotob, Ryosuke Koyamadab, Sadamu Okadab
aInternal Medicine, St. Luke’s International Hospital, Tokyo, Japan
bDivision of Hematology, St. Luke’s International Hospital, Tokyo, Japan
cCorresponding Author: Takakazu Higuchi, Division of Hematology, St. Luke’s International Hospital, 1-9 Akashi-cho, Chuo-ku, Tokyo 104-8560 Japan
Manuscript accepted for publication February 17, 2015
Short title: Acquired β-Thalassemia in Myelofibrosis
doi: http://dx.doi.org/10.14740/jh190w
| Abstract | ▴Top |
A 62-year-old man was diagnosed as primary myelofibrosis (PMF) with Janus kinase 2 V617F mutation. He also had progressive microcytic anemia which was diagnosed as acquired β-thalassemia associated with PMF. Microcytosis is reported to be complicated with approximately a quarter of the patients with PMF; however, the etiology has not been elucidated yet. To our knowledge, this is the first report of acquired β-thalassemia developed in a patient with PMF which has a clinical significance in the diagnosis and management of anemia in a subset of the patients with PMF.
Keywords: Primary myelofibrosis; Thalassemia; Microcytosis; Anemia
| Introduction | ▴Top |
Primary myelofibrosis (PMF) is a clonal stem cell disorder and frequently presents with hematological abnormalities such as leukoerythroblastosis, anemia, and thrombocytopenia of various degrees. Especially, anemia is a very common complication and anemia with hemoglobin (Hb) level below 10 g/dL is observed in 35-54% of the patients at presentation [1-3]. Thus, management of anemia is of great importance in the practice. However, PMF is not curable with conventional treatment and there is no effective therapy to ameliorate anemia partially due to the complex mechanisms [4, 5]. Reilly describes in his review that the anemia is “usually normochromic normocytic” [4]. On the other hand, Tefferi et al reported that 28% of 102 PMF patients had microcytic red blood cells (RBCs), although they did not mention the underlying mechanisms of the microcytosis [6]. Here, we report a case of acquired β-thalassemia associated with PMF as one of the mechanisms of microcytic anemia in patients with PMF and discuss the implication in the management of PMF.
| Case Report | ▴Top |
A 62-year-old man was referred to our hospital for the evaluation of anemia and splenomegaly found on the annual health check 1 month before. He was taking anti-hypertensive drugs. He had had mild normocytic anemia with the mean corpuscular volume (MCV) of 82 - 83 fL on the health checks in the previous 2 years.
On the referral, he complained of shortness of breath on exercise and night sweat. He had no specific family history. Physical examination revealed anemia, systolic heart murmur, and splenomegaly extending over the midline and the level of the navel. The complete blood count revealed that the RBC was 3.51 × 1012/L with 3.0% reticulocytes, the Hb level was 8.6 g/dL, the hematocrit was 26.3%, and the MCV was 74.9 fL. The morphology of the peripheral blood (PB) smear showed marked anisocytosis with poikilocytes, schistocytes, elliptocytes, and teardrop cells. The WBC was 4.1 × 109/L with 56.0% neutrophils, 30.5% lymphocytes, 6.5% monocytes, 2.0% metamyelocytes, 4.5% myelocytes, and 1.5% blasts and the platelet count was 98 × 109/L. The blood chemistry showed that the serum albumin level was 4.5 g/L, the creatinine was 0.79 mg/dL, the total bilirubin was 0.6 mg/dL, the asparate transaminase was 16 U/L, the alanine transaminase was 17 U/L, and the lactate dehydrogenase was 292 U/L (reference range: 118 - 223). The serum iron level was 82 μg/dL and the ferritin was 97.9 ng/mL. The HbA1c level was as low as 4.4% and the analysis of the composite of Hb by electrophoresis on cellulose acetate membrane revealed HbA of 85%, HbF of 10%, and HbA2 of 5%. The bone marrow (BM) aspiration was dry-tap and the BM biopsy revealed a marked fibrosis of grade MF-3 and increased dysplastic megakaryocytes (Fig. 1). The karyotype of the PB cells was 46,XY. Janus kinase (JAK2) V617F mutation was positive and he was diagnosed as PMF, high risk according to Dynamic International Prognostic Scoring System (DIPSS) Plus [2], and acquired β-thalassemia. Oral metenolone was started; however, pancytopenia progressed with worsening constitutional symptoms and soon he became transfusion-dependent and was transferred to another hospital in consideration of early hematopoietic stem cell transplantation.
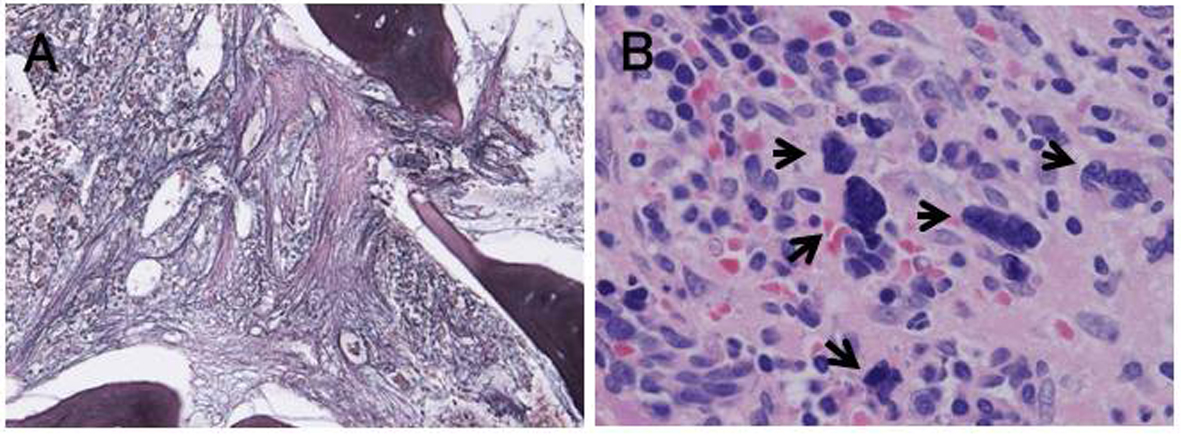 Click for large image | Figure 1. Bone marrow biopsy at diagnosis. The bone marrow was hypercellular with marked fibrosis of grade MF-3 (A, low power view, silver stain) and increased number of megakaryocytes with dysplastic features (arrows) (B, high power view, hematoxylin-eosin stain). |
| Discussion | ▴Top |
The mechanisms of anemia in PMF patients are complex and involve intrinsic erythroid cell defect, cytokine mediated inhibition, chronic hemolysis, nutritional deficiency, bleeding, hemodilution, and splenic sequestration [4, 5]. Anemia in patients with PMF is usually normocytic [4]; however, in a study at the Mayo Clinic, approximately a quarter of the patient had microcytosis of RBCs, the mechanisms of which were not studied, though [6].
Acquired thalassemia is reported to be associated with various hematological diseases, especially with myelodysplastic syndromes, and may exacerbate anemia although acquired thalassemia among PMF patients is rarely reported [7]. Acquired α-thalassemia is caused by either acquired deletion of α-globin gene cluster or inactivation mutations of trans-activating chromatin-activating factor [7]; however, acquired β-thalassemia is much rarer and its mechanism has not been fully elucidated. In case of PMF, while two cases of acquired Hb H disease associated with PMF are reported in the literature [8, 9] and increased Hb F level is found in 57% of PMF patients [10], to our knowledge, the frequency and significance of acquired β-thalassemia in PMF patients have not been reported. However, considering the high frequency of microcytosis among PMF patients [6], we believe that acquired β-thalassemia is one of the mechanisms of the microytosis and the present case exemplifies this contention. Although we could not perform genetic studies and make a definitive diagnosis, we believe that β-thalassemia in the present case is acquired rather than hereditary which developed with the progression of PMF because his RBCs were originally normocytic and he had no family history of hemoglobinopathy or anemia.
PMF is currently unable to be cured with drug therapy and patients with anemia are usually treated with androgens and the first-line therapy for splenomegaly is hydroxyurea (HU) [3]. However, the response rate of anemia to androgens is around 15-25%. Recently, a new JAK 1/2 inhibitor, ruxolitinib, is shown to be effective to reduce the spleen size and symptoms and to extend overall survival; however, it is not effective to improve anemia [11, 12]. HU is effective in ameliorating anemia in β-thalassemia patients by inducing HbF production through stress erythropoiesis. In fact, a child with hereditary β-thalassemia and myelofibrosis was treated with HU and achieved a drastic reduction of transfusion requirement [13] and HU increased HbF level in an adult patient with PMF and acquired HbH disease [9]. Although we could not treat the present case with HU and evaluate its efficacy due to the rapid progression, these patients in the literature illustrate a possibility that HU can improve anemia in PMF patients with acquired thalassemia and, together with the present case, importance to consider the possibility of acquired thalassemia in patients with microcytic anemia because HU can be effective to improve both splenomegaly and anemia in a subset of PMF patients.
Financial Support
No sources of financial support.
Conflicts of Interests
The authors have no conflict of interest to disclose.
| References | ▴Top |
- Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, Vannucchi AM, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895-2901.
doi pubmed - Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, Van Dyke D, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392-397.
doi pubmed - Tefferi A. Primary myelofibrosis: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013;88(2):142-150.
doi pubmed - Reilly JT. Pathogenesis and management of idiopathic myelofibrosis. Baillieres Clin Haematol. 1998;11(4):751-767.
doi - Guglielmelli P, Vannucchi AM. Struggling with myelofibrosis-associated anemia. Leuk Res. 2013;37(11):1429-1431.
doi pubmed - Tefferi A, Dingli D, Li CY, Mesa RA. Microcytosis in agnogenic myeloid metaplasia: prevalence and clinical correlates. Leuk Res. 2006;30(6):677-680.
doi pubmed - Steensma DP, Gibbons RJ, Higgs DR. Acquired α-thalassemia in association with myelodysplastic syndrome and other hematologic malignancies. Blood. 2005;105(2):443-452.
doi pubmed - Veer A, Kosciolek BA, Bauman AW, Rowley PT. Acquired hemoglobin H disease in idiopathic myelofibrosis. Am J Hematol. 1979;6(3):199-206.
doi pubmed - Rinaldi CR, Rinaldi P, Pane F, Camera A, Rinaldi C. Acquired Hb H disease associated with elevated Hb F level in patient affected by primary myelofibrosis. Ann Hematol. 2010;89(8):827-828.
doi pubmed - Mendek-Czajkowska E, Slomkowski M, Zdebska E, Mokras U, Sikorska A, Maryniak R, Gorski T, et al. Hemoglobin F in primary myelofibrosis and in myelodysplasia. Clin Lab Haematol. 2003;25(5):289-292.
doi pubmed - Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-I. Haematologica. 2013;98(12):1865-1871.
doi pubmed - Cervantes F, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Sirulnik A, Stalbovskaya V, McQuitty M, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047-4053.
doi pubmed - Nirupam N, Maheshwari A, Rath B, Chandra J, Kumar P, Basu S, Nangia A. Myelofibrosis: a cause of increased transfusion requirement in a child with β-thalassemia intermedia. J Pediatr Hematol Oncol. 2012;34(2):143-145.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.