

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 4, Number 3, September 2015, pages 205-209
A Case of Evans Syndrome: A Clinical Condition With Under-Recognized Thrombotic Risk
Zachary D. Otaibia, c, Rohit Raob, Santhosh K. Sadashivb
aDepartment of Internal Medicine, Allegheny Health Network, USA
bDepartment of Hematology and Oncology, Allegheny Health Network, USA
cCorresponding Author: Zachary D. Otaibi, Department of Internal Medicine, Allegheny Health Network, USA
Manuscript accepted for publication August 18, 2015
Short title: Thrombotic Risk in Evans Syndrome
doi: http://dx.doi.org/10.14740/jh222w
| Abstract | ▴Top |
Evans syndrome (ES) is a rare hematologic disorder characterized by the presence of Direct Antiglobulin test (DAT) positive autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP), and/or immune neutropenia. The risk of thrombosis has been well established in retrospective studies for ITP and AIHA; however, the risk of thrombosis in ES has been limited to case reports and individual case series. Whether the risk of thrombosis in ITP and AIHA is additive has not been well established, but preliminary observation suggests the rate of thrombosis is higher than the rate observed in ITP or AIHA individually. Furthermore, ES appears to be an underappreciated diagnosis. Anemia in the presence of ITP is often considered to be secondary to acute blood loss and a proper workup for hemolysis is often missed. Appropriate risk stratification of these patients is hindered by this lack of workup and many patients are unfortunately not treated appropriately and/or not offered appropriate prophylaxis for their level of thrombotic risk. The purpose of this case report is to not only increase the level of awareness among clinicians to initiate an appropriate diagnostic workup in patients presenting with anemia in the setting of ITP, but also to a heightened thrombotic risk warranting an appropriate thromboprophylaxis. This case also adds to the body of evidence that a “second- hit” phenomenon is often a precipitating cause for a thrombotic event.
Keywords: Evans syndrome; Thrombosis; May Thurner; Lupus anticoagulant; Antiphospholipid antibodies; DVT
| Introduction | ▴Top |
Evans syndrome (ES) is a hematologic disorder characterized by the sequential or simultaneous development of DAT positive autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP) and/or immune neutropenia in the absence of a known etiology. The syndrome was first described by Robert Evans in 1951 when he demonstrated a relationship between primary immune thrombocytopenia and acquired hemolytic anemia [1]. Since this description, much of our current understanding of ES is derived through case studies and individual case reports. In 2005, the European Hematology Association’s working group on thrombocytopenia set out to define the clinical spectrum of ES and describe its clinical features. In their retrospective analysis of 68 cases of ES, half of the reviewed cases (34) were considered primary ES and half were associated with an underlying autoimmune, infectious, lymphoproliferative, or immunodeficiency related disorder. They also concluded ES, and its associated conditions, can be described as a state of profound immune dysregulation [2].
Thrombosis as a complication of ITP and AIHA has been well documented in medical literature; however, there are a limited number of ES cases which report thrombotic complications. One case published in 1994 provides a description of a 24-year-old male diagnosed with ES and recurrent deep vein thrombosis (DVT). This patient met the American College of Rheumatology’s (ACR) 1982 classification criteria for systemic lupus erythematosus (SLE), and furthermore, the patient was positive for antiphospholipid antibodies (APLAs). This patient ultimately required above-knee amputation despite prompt recognition and treatment [3]. In 2005, a case of ES and cerebral venous thrombosis was reported in a 19-year-old male who presented with complaints of headache, convulsive seizure, and vomiting. This patient tested negative for APLA but was found to have prothrombin G20210A gene mutation predisposing him to thrombosis [4]. Lastly, in a case series published in 2009, two patients with primary antiphospholipid syndrome (APS) and ES were described. The first patient was a 19-year-old female who presented with acute lower extremity DVT and the other patient was a 36-year-old female with recurrent fetal loss and epistaxis. Both of these patients were treated with intravenous (IV) steroids and had reported resolution of their cytopenias [5]. Each of the unique cases outlined above demonstrates patients meeting the diagnostic criteria for ES, but more importantly, they shared a common feature of a second-hit which increased their risk for thrombosis [6].
In this case report, we describe a 63-year-old female with a long standing history of ES who developed an acute lower extremity DVT after aggressive treatment of her thrombocytopenia. With the initial diagnosis of ITP made, the presence of AIHA was not recognized as a part of her clinical presentation and the presence of AIHA was not established until late in her treatment course. This patient had multiple risk factors for thrombosis including positive APLAs and possible May Thurner anatomy. To our knowledge, this is the only case of venous thrombosis in the setting of ES and May Thurner anatomy to be reported. This case also adds to the body of evidence that thrombosis in the setting of ES often has a precipitating “second hit” component.
| Case Report | ▴Top |
A 63-year-old female was transferred to our facility with a history of extensive DVT with clinical signs of phlegmasia cerulea dolens. Her only significant past medical history was ES, diagnosed more than 20 years ago that was managed with intermittent steroids during the flare-ups, but eventually required splenectomy which resulted in long term remission lasting more than a decade. She presented to an outside hospital with complaints of upper and lower extremity petechiae, hematuria, and mucosal bleeding (wet purpura). Following clinical evaluation and with CBC revealing a WBC count of 5.81 × 103/μL, Hb of 13.3 g/dL and a platelet count of 3.0 × 103/μL, ITP was suspected. She was administered IV steroids, intravenous immunoglobulin (IVIG), and platelet transfusions. Despite several days of treatment with this regimen, she did not demonstrate platelet recovery and given continued hematuria, second line therapy with danazole followed by romiplostim was initiated. Three days after initiating second line therapy, she reported acute pain and swelling in her left lower extremity. Doppler studies revealed an extensive DVT involving the left lower extremity and the patient had clinical signs consistent with phlegmasia cerulea dolens. The patient was transferred for higher level of care given the degree of clot burden in the setting of thrombocytopenia.
The patient on presentation at the tertiary care facility had a cold left lower extremity with associated 2+ pitting edema. Her CBC upon arrival was significant for a WBC 25.86 × 103/μL, Hb 9.0 g/dL and platelet count 45 × 103/μL (Table 1). Computed tomography (CT) of the abdomen, pelvis, and bilateral lower extremities demonstrated a filling defect of the left common iliac, internal and external iliac, left common and superficial femoral veins as well as poor filling of the left popliteal and tibial veins. Also noted was possible compression of the left common iliac vein by the right common iliac artery concerning with May Thurner syndrome (Fig. 1). Computed tomography angiography (CTA) of the chest excluded the possibility of pulmonary embolism. Hypercoagulable workup revealed positive lupus anticoagulant (LA), anticardiolipin antibodies, antiphosphatidylserine antibodies and B2-glycoprotein antibodies (Table 2). The patient’s direct antiglobulin test (DAT) was positive (IgG 4+). Peripheral smear was positive for spherocytes and the patient’s reticulocyte count was elevated at 8.7%. ANA, IFA was positive with a 1:320 diffuse, homogenous pattern, but anti-DS DNA was negative. Given a positive DAT and presence of thrombocytopenia, she was diagnosed with ES.
 Click to view | Table 1. Initial and Follow-Up Labs After 12-Weeks: CBC, Reticulocyte Count, Hemolysis Labs and ANA |
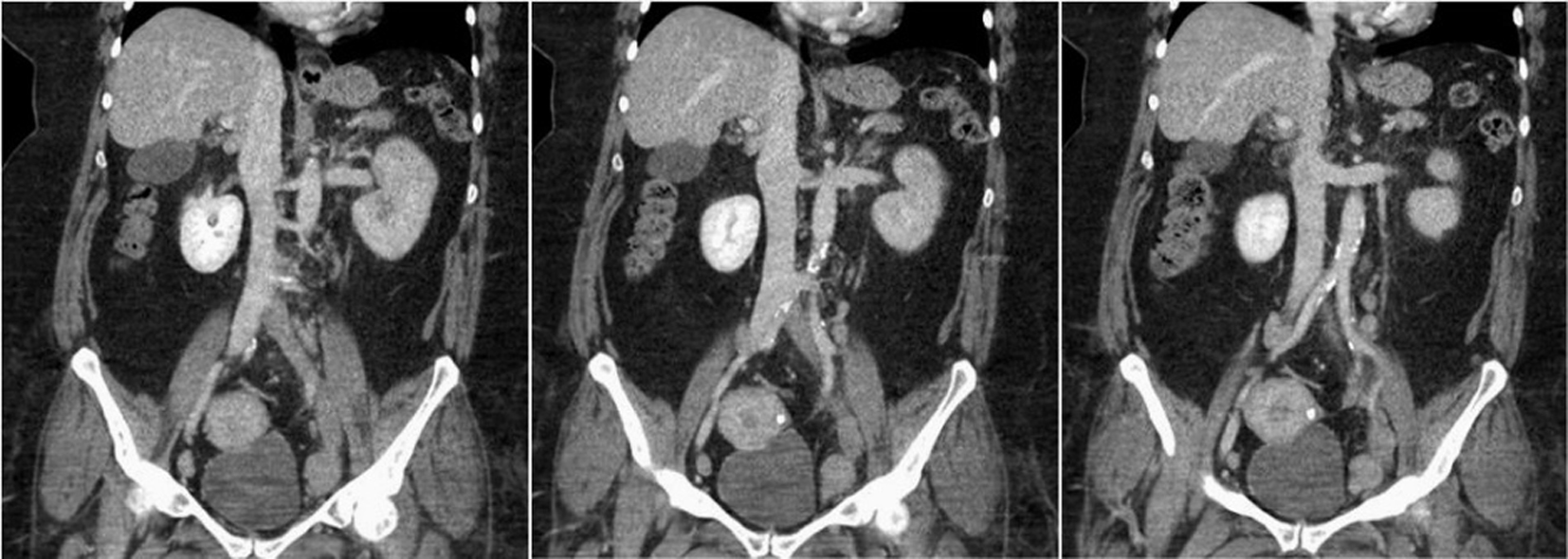 Click for large image | Figure 1. CT of abdomen/pelvis: filling defect/lack of opacification of the left common iliac, internal and external iliac with apparent compression of the left common iliac vein by the right common iliac artery. Findings are most consistent with May Thurner syndrome. |
Click to view | Table 2. Initial and 12-Week Follow-Up APLA Testing |
Decision making and management
Following diagnosis of ES, she was started prednisone at a dose of 1 mg/kg body weight daily. The presence of hematuria along with extensive DVT prompted platelet transfusion to achieve a platelet count of ≥ 50,000/µL while anticoagulation was initiated with IV heparin. Catheter directed thrombolysis (CDT) was deemed high risk given her active hematuria and hence was not recommended. The patient was maintained on heparin with recovery of her platelet counts and improvement in her left lower extremity symptoms and hematuria. Following improvement in both clinical and hematological parameters, the prednisone dose was tapered and anticoagulation was switched to full dose subcutaneous enoxaparin 1 mg/kg body weight twice daily. As she continued to make recovery, she was discharged on oral prednisone and full dose enoxaparin as a bridge to warfarin. On the day of discharge, the patient’s platelet count was 190,000/µL and hemoglobin was 9.1 g/dL.
| Discussion | ▴Top |
In ES, the AIHA is largely the warm-agglutinin subtype. This subtype represents 80% of all cases of AIHA and is characterized by autoimmune destruction of red blood cells (RBCs). Antibodies, predominantly of the IgG subclass, affix themselves to RBC surface antigens forming an RBC-IgG complex [7]. This complex directly interacts with the Fc receptors located on cytotoxic cell surfaces. For phagocytosis to ensue multiple RBC-IgG-Fc interactions must occur. The ideal location for these interactions to occur is located in the sinusoids of the spleen resulting in splenic sequestration and splenomegaly. RBCs in which there is incomplete or partial phagocytosis develop an increased surface to volume ratio becoming spherocytes [8, 9]. Review of peripheral smear for presence of spherocytes can provide valuable clues in addition to laboratory findings of elevated lactate dehydrogenase (LDH), low haptoglobin, reticulocytoses and elevated indirect bilirubin. However, the gold standard for diagnosing remains the DAT. This test is positive in 99% of patients with warm-type AIHA [10].
The antibodies seen in ES and ITP are different than the autoantibodies present in AIHA. The antibodies in ITP are IgG autoantibodies and are directed at platelet derived glycoproteins; one of the most common targets is the GPIIb/IIIa glycoprotein. These antibodies lead to premature destruction and a reduction in platelet lifespan [11]. The pathogenesis and development of these antibodies is poorly understood; however, both genetic and acquired mechanisms are believed to contribute. A number of infectious and systemic diseases have been linked to the formation of autoantibodies and the development of this clinical syndrome [12, 13]. However, unlike AHIA, there are no assays available to directly aid in the diagnosis. Rather, ITP remains a diagnosis of exclusion and relies on a detailed history, physical examination, and lab work excluding other causes of thrombocytopenia.
Although proper identification of AIHA and ITP is essential for diagnosing ES, it is even more crucial for risk stratifying patients for potential complications. Both ITP and AIHA have been linked with increased rates of arterial and venous thrombosis. Whether the individual risks are additive in patients with ES has not been well established, but observation suggests the risk for venous thromboembolism (VTE) and arterial clots is higher in patient with ES than with ITP or AIHA alone.
A study of ITP from the UK general practice database including 1,070 adult patients diagnosed with ITP concluded there was a higher risk of venous and arterial thrombosis with a reported hazard ratio (HR) of 1.58 (95% CI: 1.01 - 2.48) and 1.43 (95% CI: 1.02 - 2.02), respectively [14]. In another large-scale study completed in 2014, Yousef et al reviewed insurance claims submitted from a large-scale private insurance company from 2007 to 2010. A total of 2,772 patients held the diagnosis of ITP. Patients were followed for a period of 4 years and adjusted HR for VTE was calculated. HRs were 5.50, 4.49, 2.95, 1.29, 0.57, and 0.25 at 90 days, 120 days, 1 year, 2 years, 3 years, and 4 years, respectively. The same analysis was completed for AIHA. Again, the HR for VTE was significantly elevated in this cohort and even higher than the ITP subgroup [15]. Many additional studies over the past several decades have confirmed these findings and have provided similar conclusions.
The pathogenesis of thrombosis in ES and its related disorders remain poorly defined, but several mechanisms have been proposed. Pro-inflammatory cytokines, cell-derived microparticles, and direct endothelial injury caused by platelet and RBC-directed surface antibodies are all believed to contribute to the pathogenesis and increased risk of thrombosis in ITP and AIHA. Beyond these mechanisms, treatment directed towards AIHA , ITP and ES adds to the thrombotic risk. Higher rates of thrombosis have been linked with the use of oral steroids, IVIG, thrombopoientin-receptor agonists (TPO-R), and splenectomy.
Among these patients, it is not uncommon to find additional risk factors to be present (“second-hit”). Many patients will have systemic diseases present such as SLE or lymphoproliferative disorders. A detectable level of APLAs is often found in patients with ES and ITP. Additionally, coagulation cascade disorders and prothrombin gene mutations have been linked in medical literature.
In our case, despite a history of ES, anemia was interpreted as a consequence of bleeding due to ITP rather than an AIHA process. This assumption by treating clinicians is not infrequent and can lead to incorrect diagnoses and treatment that may result in serious complications, as illustrated above. In addition, presence of APLAs, LA, and possibility of an underlying May Thurner’s syndrome supports the “second-hit” hypothesis that has been described to significantly increase the risk for VTE in such patients [6]. Although screenings for APLA, genetic mutations, and anatomical abnormalities are not recommended for risk stratification, the proper workup of anemia when present should not be overlooked in patients presenting with thrombocytopenia and should be evaluated for an underlying AIHA. Despite the evidence of heightened prothrombotic state in patients with ES (ITP + AIHA), there are no established guidelines for thromboprophylaxis. Studies have suggested that presence of LA in patients with AIHA are at significantly higher risk for VTE and some institutions have adopted LA as a predictive marker for risk stratification to recommend an appropriate anticoagulation. In addition given the rarity of ES, treating physicians may lack the understanding of the underlying pathophysiological factors contributing to a heightened prothrombotic risk. These factors may result in inadequate thromboprophylactic treatment leaving these patients vulnerable to a thrombotic event. We hope that our case report will trigger further investigation to identify core clinical and laboratory findings that will help in stratification of patients with diagnosis of ES for risk of VTE.
Conflicts of Interest
All authors have no conflicts of interest to disclose.
| References | ▴Top |
- Evans RS, Takahashi K, Duane RT, Payne R, Liu C. Primary thrombocytopenic purpura and acquired hemolytic anemia; evidence for a common etiology. AMA Arch Intern Med. 1951;87(1):48-65.
doi pubmed - Michel M, Chanet V, Dechartres A, Morin AS, Piette JC, Cirasino L, Emilia G, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114(15):3167-3172.
doi pubmed - Al-Fiar FZ, Clink HM. Evans syndrome associated with venous thrombosis. Ann Saudi Med. 1995;15(2):168-170.
pubmed - Yilmaz S, Oren H, Irken G, Turker M, Yilmaz E, Ada E. Cerebral venous thrombosis in a patient with Evans syndrome: a rare association. Ann Hematol. 2005;84(2):124-126.
doi pubmed - Khalifa M, Ghannouchi N, Kaabia N, BenJazia E, Hachfi W, Krifa A, Letaief A, et al. Primary antiphospholipid syndrome and Evan's syndrome: 2 case reports. Acta Clin Belg. 2009;64(1):65-67.
doi pubmed - Kim KJ, Baek IW, Yoon CH, Kim WU, Cho CS. Thrombotic risk in patients with im-mune thrombocytopenia and its association with antiphospholipid antibodies. Br J Haematol. 2013;161(5):706-714.
doi pubmed - Packman CH. Hemolytic anemia due to warm autoantibodies. Blood Rev. 2008;22(1):17-31.
doi pubmed - Alwar V, Shanthala DA, Sitalakshmi S, Karuna RK. Clinical patterns and hematological spectrum in autoimmune hemolytic anemia. J Lab Physicians. 2010;2(1):17-20.
doi pubmed - Semple JW, Freedman J. Autoimmune pathogenesis and autoimmune hemolytic anemia. Semin Hematol. 2005;42(3):122-130.
doi - Segel GB, Lichtman MA. Direct antiglobulin ("Coombs") test-negative autoimmune he-molytic anemia: a review. Blood Cells Mol Dis. 2014;52(4):152-160.
doi pubmed - Chan H, Moore JC, Finch CN, Warkentin TE, Kelton JG. The IgG subclasses of platelet-associated autoantibodies directed against platelet glycoproteins IIb/IIIa in patients with idio-pathic thrombocytopenic purpura. Br J Haematol. 2003;122(5):818-824.
doi pubmed - Ahn YS, Horstman LL. Idiopathic thrombocytopenic purpura: pathophysiology and man-agement. Int J Hematol. 2002;76(Suppl 2):123-131.
doi pubmed - Pullarkat V, Ngo M, Iqbal S, Espina B, Liebman HA. Detection of lupus anticoagulant identifies patients with autoimmune haemolytic anaemia at increased risk for venous thrombo-embolism. Br J Haematol. 2002;118(4):1166-1169.
doi pubmed - Sarpatwari A, Bennett D, Logie JW, Shukla A, Beach KJ, Newland AC, Sanderson S, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica. 2010;95(7):1167-1175.
doi pubmed - Yusuf HR, Hooper WC, Grosse SD, Parker CS, Boulet SL, Ortel TL. Risk of venous thromboembolism occurrence among adults with selected autoimmune diseases: a study among a U.S. cohort of commercial insurance enrollees. Thromb Res. 2015;135(1):50-57.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.