


| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 3, Number 1, March 2014, pages 13-18
Erythroblastic Synartesis in a Patient With Low Grade B Cell Lymphoma
Kevin O. Schmidta, Jonathan E. Dowella, Anant Sharmaa, Daniel R. Nussenzveigc, d, David H. Wanga, Henrik Illuma, e
aDepartment of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX, USA
bMedical Service, Veterans Affairs North Texas Health Care System, Dallas, TX, USA
cDepartment of Pathology, University of Texas Southwestern Medical Center, Dallas, TX, USA
dPathology and Laboratory Medicine Service, Veterans Affairs North Texas Health Care System, Dallas, Texas 75216, USA
eCorresponding author: Henrik Illum, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX, USA
Manuscript accepted for publication December 4, 2013
Short title: Erythroblastic Synartesis
doi: https://doi.org/10.14740/jh113w
| Abstract | ▴Top |
Erythroblastic synartesis is a very rare form of acquired dyserythropoiesis. It is characterized by ineffective erythropoiesis due to abnormal aggregates of erythroid precursors in the bone marrow. An autoimmune etiology has been implicated with several cases having an IgG kappa paraprotein. The first case was described in 1973 and a total of 9 cases have been reported to date. There appears to be an association with lymphoproliferative and autoimmune disorders and treatment with steroids, rituximab and other immunosuppressants have been utilized with varying degrees of benefit. We here describe a new case of erythroblastic synartesis in a 59-year African-American man who presented with an IgG kappa paraprotein and an associated low grade B-cell lymphoma. The condition in this patient has proved relatively treatment refractory over the course of almost four years, necessitating multiple sequential lines of therapy with steroids, rituximab, erythropoietin and cyclosporine. A review of the previously published reports will be provided in addition to the current case presentation.
Keywords: Erythroblastic synartesis; Anemia; Dyserythropoesis; Rituximab; Prednisone; Cyclosporine; Low grade B-cell lymphoma
| Introduction | ▴Top |
Erythroblastic synartesis is a very rare form of acquired dyserythropoiesis characterized clinically by an often severe normocytic normochromicanemia with marked reticulocytopenia. The disorder was first described by Gorius et al in 1973 in a patient who presented with a two years history of chronic anemia [1]. Bone marrow aspiration revealed marked erythroid hyperplasia with a large proportion of erythroblasts being bi-nucleated or multi-nucleated. The most striking feature noted was the presence of clumps of erythroblasts of various ages without obvious cytoplasmic separation, giving them a syncytial appearance. Electron microscopy demonstrated the erythroblasts being linked together with interdigitatingcell processes comprised of regularly spaced septate-like structures devoid of ferritin. The term erythroblastic synartesis (from Greek “with link”) was later suggested by the authors in 1974 [2]. Similar morphologic observations were made in 1976 in a Japanese patient that presented with an unexplained dyserythropoieticanemia [3].
The largest case series to date was reported by Cramer et al in 1999 [4]. A total of three patients who presented over a period of 22 years were described. They all had findings on bone marrow aspirate and electron microscopy that closely mirrored the first two published reports. The erythroblastic synartesis could further be reproduced in vitro, using bone marrow progenitor cells from two of the patients. Crossed cell culture experiments demonstrated presence of erythroblastic synartesis when autologous serum was used for incubation. This observation was not made when control serum was utilized. They further noted that erythroblastic synartesis was absent when IgG depleted autologous patient serum was used for incubation, suggesting that an IgG component was causally responsible for the observations. Using immuno-electron microscopy they were able to demonstrate positive immunogold labeling for immunoglobulin gamma chains specifically along the synartesis. Immunolabeling for immunoglobulin light chains demonstrated only kappa light chains at the site of the synartesis. Control labeling on normal erythroid islands was reported as consistently being negative for both gamma and kappa chains. Immunodetection for the adhesion receptor CD36 revealed labeling in the areas of synartesis, where it was found to be concentrated compared to the uninvolved erythroblast surface, suggesting it as a possible responsible target antigen.
An additional 4 cases have since been reported [5-8]. Zaninoni et al performed bone marrow cultures on a patient who presented with clinical and morphological changes compatible with erythroblastic synartesis [5]. They reported the mitogen-stimulated direct antiglobulin test as being strongly positive, supporting an autoimmune etiology. The patient’s bone marrow supernatant was in addition able to induce typical morphological changes of erythroblastic synartesis in normal bone marrow.
The clinically salient features of the published reports to date have been summarized in Table 1. An IgG kappa monoclonal paraprotein was reported in four cases, was not found in one case and was not commented on in the other four patients. Underlying lymphoproliferative disease was reported in three patients, two with small lymphocytic lymphoma (SLL) and one with chronic lymphocytic leukemia (CLL). One patient was diagnosed with Sjogrens syndrome and two patients had no apparent associated underlying diseases found after an extensive workup. In the remaining three cases, there was no mention of an evaluation for an additional hematological disorder. When commented on, patients appeared to have responded generally quite well to treatment with steroids and in some cases other immunosuppressive therapies (e.g. rituximab, CHOP, fludarabine and cyclosporine).
 Click to view | Table 1. The Clinically Salient Features of the Published Reports |
| Case Report | ▴Top |
We report here a 59-year-old African-American male with unremarkable prior medical history and hemogram, who presented with a two week history of fatigue, generalized weakness and shortness of breath. A complete blood count revealed a hemoglobin of 4.7 g/dL (MCV 70), leukocyte count of 14.6 K/cu mm (normal differential), and a normal platelet count. Clinical history for drug and toxin exposures was unrevealing. The LDH was 532 U/L, total bilirubin 1.9 mg/dL, direct bilirubin 0.3 mg/dL, haptoglobin < 15 mg/dL, ferritin 273 ng/dL, vitamin B12 479 pg/dL, serum folate 6.6 ng/ml and reticulocyte count 0.9%. Serologic testing for HIV, hepatitis B and C was negative. Serum protein electrophoresis was remarkable for two IgG kappa monoclonal paraproteins (0.15 g/dL and 0.79 g/dL).
Peripheral smear showed severe microcytic, hypochromic anemia with marked poikilocytosis and numerous nucleated red blood cells with dysplastic changes such as bi-nucleation, karyorrhexis, nuclear irregularities, nuclear budding and coarse basophilic stippling. Most of the circulating nucleated red blood cells were in the orthochromatic or late polychromatophilicstage of maturation, but rare earlier precursors, such as early basophilic or even proerythroblasts, were also seen. A small monotonous population of small mature lymphocytes occasionally exhibiting polar cytoplasmic projections was also noted (Fig.1). The other cell lines were unremarkable.
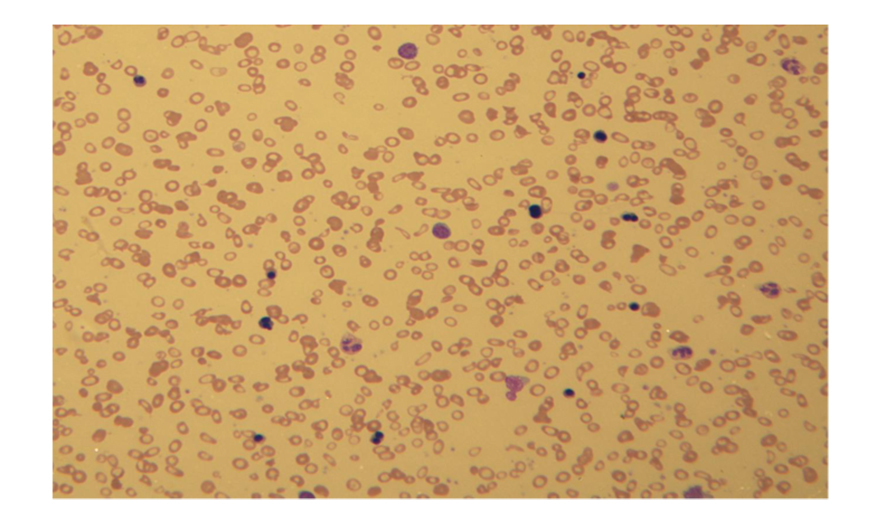 Click for large image | Figure 1. Severe microcytic, hypochromic anemia with marked poikilocytosis and numerous nucleated red blood cells with dysplastic changes including bi-nucleation. |
The patient's bone marrow biopsy and aspirate was dominated by striking erythroid hyperplasia with multiple confluent islands of erythroid precursors displaying progressive maturation with obvious dyserythropoiesis, most noticeable by the presence of bi- or multi-nucleation and karyorrhexis.
Many of the proerythroblasts and less mature precursors appeared to cluster together in small clusters (syncytia) with close apposition of their plasma membranes, joined at their nonbasophilic cytoplasmic clear zone, where they appeared flattened with a tile-like pattern (synartesis) (Fig 2, 3).
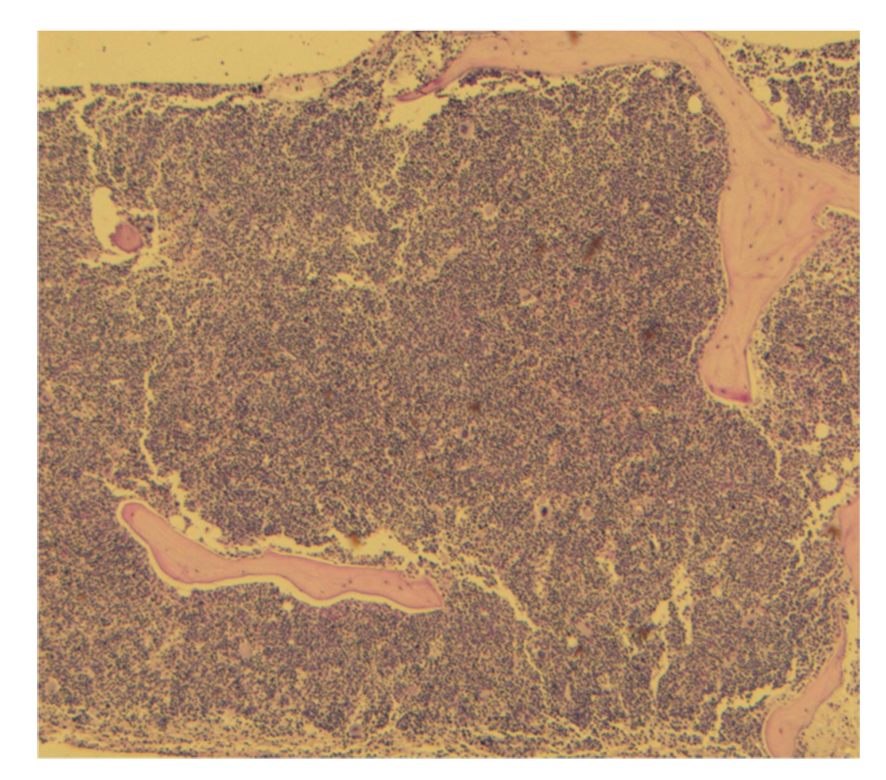 Click for large image | Figure 2. Strikingly hypercellular bone marrow biopsy. |
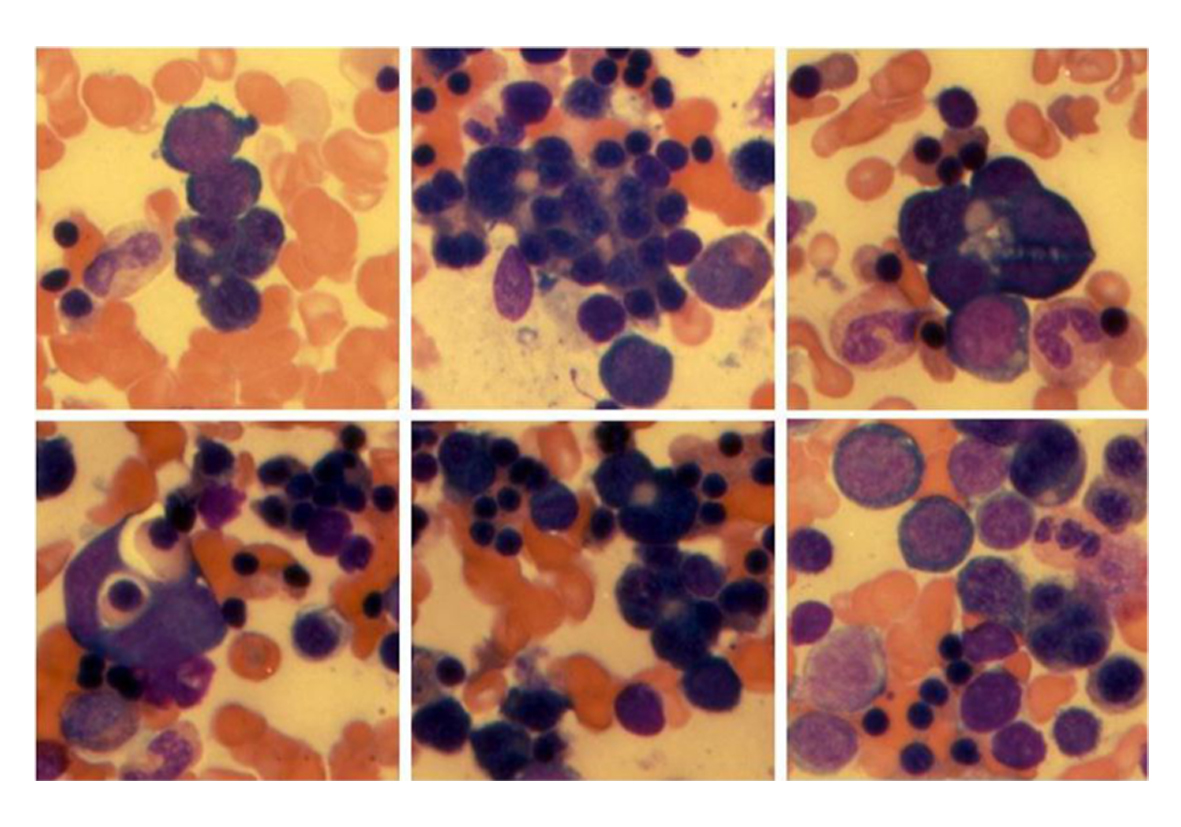 Click for large image | Figure 3. Multiple examples of erythroblastic synartesis or syncytial-like cluster of erythroblasts linked together by close apposition of their plasma membranes forming a nonbasophilic cytoplasmic clear zone. There is extreme erythroid hyperplasia with marked dyserythropoiesis (unilineage dysplasia). |
The other cell lines were preserved except for a mild somewhat diffuse increase in the number of lymphoid cells with the formation of small, predominantly paratrabecular aggregates. Flow cytometric analysis revealed a 5-7% population of small sized cells with the immunophenotype: CD19 (+), CD20 (dimly +), CD5 (dimly +), CD10 (-), CD11c (variably +), CD22 (dimly +) and CD23 (-). Cytogenetic studies with conventional karyotyping and a MDS panel by fluorescence in situ hybridization (FISH) were negative.
Computed tomography of the chest, abdomen and pelvis was unremarkable except for widespread low volume (mostly subcentimeter) lymphadenopathy involving supraclavicular, axillary mediastinal, hilar and pelvic nodal stations. The largest lymph node measured 1.6 cm and was not appreciated on physical exam. With these findings, the overall impression was that of erythroblastic synartesis with an associated low grade B cell lymphoma/lymphoproliferative disorder, most likely splenic marginal zone lymphoma.
The initial treatment consisted of dexamethasone (40 mg PO for 4 days) and weekly rituximab (375 mg/m2 for 4 weeks). Despite this and a total of 4 blood transfusions (7 units) in the three months after his diagnosis, the patient's hemoglobin did not stabilize. He was subsequently started on erythropoietin (40,000 units per week) which maintained his hemoglobin above 9 g/dL, but eight months after beginning erythropoietin therapy, the patient's anemia began to worsen and he became transfusion dependent. Steroids were started and the erythropoietin dose was increased to 60,000 units per week. The hemoglobin improved and almost normalized for about 1 - 2 months. Worsening anemia led to cessation of steroids and another trial of rituximab which provided a good response lasting for about 3 - 4 months. The patient again developed transfusion dependence prompting a third trial of rituximab, which unfortunately provided little to no benefit. The patient was restarted on steroids (60 mg prednisone daily), and he experienced a good response with hemoglobin levels as high as 11.8 g/dL. Attempts to taper oral steroids resulted in relapse with declining hemoglobin levels. The patient was therefore started on cyclosporine as a steroid sparing therapy. He has thereafter maintained his hemoglobin levels at approximately 10 g/dL and steroid taper was resumed down to 20 mg of prednisone per day. The treatment course has been summarized in Figure 4.
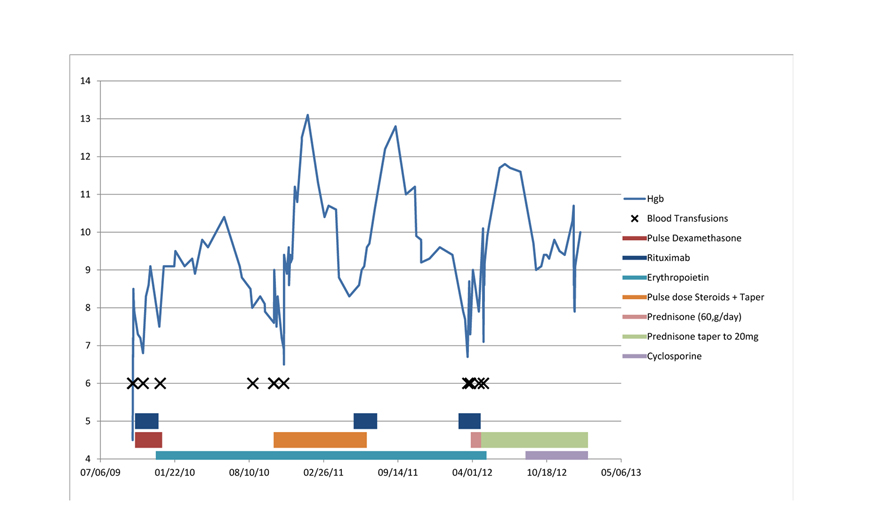 Click for large image | Figure 4. The treatment course. |
The two IgG kappa monoclonal paraproteins have persisted over time but the levels have correlated inversely with the hemoglobin. Another feature in this patient, not previously described, was the microcytic and hypochromic appearance of his erythrocytes in the peripheral blood. There was no laboratory evidence for iron deficiency. These unusual morphologic features have also correlated with the degree of anemia, with improvement during periods of higher hemoglobin.
Conclusions
We describe here what we believe is the 10th published case report of erythroblastic synartesis. The dyserythropoetic morphological features in the bone marrow were very similar to those previously reported. In addition, this patient also had an underlying low grade B-cell lymphoma with overall features not definitively conclusive for, but mostly suggestive of, splenic marginal zone lymphoma. This further strengthens the previous association with indolent B-cell lymphoproliferative diseases and erythroblastic synartesis, with two prior reports of concurrent small lymphocytic lymphoma and one patient with chronic lymphocytic leukemia. The work presented by Cramer et al and Zaninoni et al have strongly implicated an underlying autoimmune etiology most likely mediated by an IgG kappa paraprotein, which was present in all but one of the prior cases in which an evaluation for a paraprotein was undertaken. Our patient had two small IgG kappa monoclonal paraproteins on presentation, which have persisted, but with levels correlating inversely with the degree of anemia over time. This is the first report of two paraproteins being associated with erythroblastic synartesis, raising the question about which one might be responsible for the dyserythropoesis. An additional unique feature of the present case was the microcytic and hypochromic appearance of the erythrocytes in the peripheral blood smear. The observed correlation between these findings and the degree of anemia along with the absence of evidence of iron deficiency, suggests that these changes in red cell morphology likely represents a variant presentation of erythroblastic synartesis.
The previous reports have described generally quite favorable responses to treatment with steroids, rituximab and other immunosuppressants. This has unfortunately not been our experience with this patient who has had a rather treatment refractory course over almost four years, necessitating multiple sequential lines of therapy with steroids, rituximab, erythropoietin and cyclosporine. During this time, the underlying low grade B-cell lymphoma has shown no evidence of progression or transformation. It is our hope that this report and literature review will further add to the limited knowledge base of this rare and likely under-recognized condition.
Competing Interests
None of the authors have any competing interests to disclose.
| References | ▴Top |
- Gorius JB, Flandrin G, Daniel MT, Brouet JC. Septate-like junctions acquired by erythroblasts in a case of refractory anaemia. Scand J Haematol. 1973;10(3):219-224.
doi pubmed - Breton-Gorius JD, MT, Guichard J, Flandrin G. Cr Acad Sci Paris. 1974;278:73.
- Tani N, Harasawa S, Suzuki S, Miwa M, Nomiyama T, Senoue I, Kikuchi K, et al. Gastric acid secretion and plasma gastrin response to test meal in patients with gastric cancer. Tokai J Exp Clin Med. 1982;7(2):159-172.
pubmed - Cramer EM, Garcia I, Masse JM, Zini JM, Lambin P, Oksenhendler E, Souni F, et al. Erythroblastic synartesis: an auto-immune dyserythropoiesis. Blood. 1999;94(11):3683-3693.
pubmed - Zaninoni A, Imperiali FG, Pomati M, Colombi M, Boschetti C, Barcellini W. Bone marrow mitogen-stimulated direct antiglobulin test in a case of erythroblastic synartesis. Clin Lab. 2010;56(9-10):459-462.
pubmed - Bacher U, Lenz G, Haferlach T, Loeffler H, Moosmann N, Hiddemann W, Ostermann H. Erythroblastic synartesis in a patient initially diagnosed with myelodysplastic syndrome. Ann Hematol. 2005;84(4):272-273.
doi pubmed - Liapis K, Bakiri M, Apostolidis J. Erythroblastic synartesis in a patient with small lymphocytic lymphoma. Br J Haematol. 2008;142(1):2.
doi pubmed - Papakonstantinou G, Loeffler H, Haferlach T, Brugger W. Severe idiopathic erythroblastic synartesis: successful treatment with the anti-CD20 monoclonal antibody rituximab. Eur J Haematol. 2010;84(6):547-549.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.