

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Case Report
Volume 10, Number 3, June 2021, pages 143-146

Schnitzler’s Syndrome: A Diagnostic Consideration in Evaluating the Constellation of Monoclonal Gammopathy and Chronic Urticaria
Jyothika Mamadgia, b, Laila Babara, Rama Bhagavatulaa, Santhosh Sadashiva, Kossivi Danteya, Mariya Apostolovaa
aAllegheny Health Network Cancer Institute, Pittsburgh, PA, USA
bCorresponding Author: Jyothika Mamadgi, Allegheny Health Network Cancer Institute, Pittsburgh, PA, USA
Manuscript submitted February 19, 2021, accepted April 1, 2021, published online June 16, 2021
Short title: A Case of Schnitzler’s Syndrome
doi: https://doi.org/10.14740/jh800
| Abstract | ▴Top |
Schnitzler’s syndrome is a rare clinical entity characterized by intermittent, non-pruritic urticarial rash, fevers, arthralgias, myalgias and monoclonal gammopathy, most commonly of the immunoglobulin M (IgM) subtype. Schnitzler’s syndrome should be considered in the differential diagnosis of fever of unknown origin. We report a case of a 56-year-old healthy Caucasian female, who initially presented to the primary care physician’s office with complaints of severe generalized fatigue and myalgias involving thighs and calves. Patient subsequently underwent extensive rheumatologic workup, and was treated with multiple courses of steroids with temporary resolution of symptoms. During the course of her workup she was found to have IgM kappa monoclonal gammopathy, and was referred to hematology for further evaluation. The constellation findings of fever, arthralgias, chronic intermittent non-pruritic urticaria, myalgias, and a negative rheumatologic workup in the presence of IgM monoclonal gammopathy raised the suspicion of Schnitzler’s syndrome. Following completion of additional workup, she was started on anakinra 100 mg daily with prompt resolution of her symptoms. Due to the rarity of the disease, the diagnosis of Schnitzler’s syndrome is often delayed, with an average time to diagnosis being approximately 5 years. The symptoms in most cases can be debilitating and add to significant morbidity as noted in our patient, who required bilateral hip arthroplasty at a much younger age than expected. Published reports discuss the poor quality of life associated with the delayed diagnosis and unawareness of potential end organ damage. With our case report we like to highlight the disease characteristics for an early identification to prevent further organ damage believed to be from chronic inflammation. Early diagnosis and treatment with agents such as interleukin-1 (IL-1) inhibitors can promptly provide symptomatic relief, reduce inflammation and prevent organ damage.
Keywords: Schnitzler’s syndrome; IgM monoclonal gammopathy; Chronic urticaria; Inflammatory syndrome; Anakinra; IL-1 receptor antagonist; Fever of unknown origin
| Introduction | ▴Top |
Schnitzler’s syndrome is a rare auto inflammatory disease characterized by symptoms of intermittent non-pruritic urticaria, fever, arthralgia and monoclonal gammopathy, all mediated by overproduction of interleukin-1 (IL-1) [1]. The syndrome was first officially reported in 1974, by a French dermatologist, Dr. Schnitzler [1, 2]. Less than 300 cases have been reported worldwide [1]. Schnitzler’s syndrome affects adults usually in their 50s [1, 2]. The diagnosis is difficult to establish and is often delayed, with an average time to diagnosis being approximately 5 years [3], adding significant morbidity to the affected patients. Multiple attempts have been made to simplify the diagnostic process. One such criterion defined by Simon et al [1], the Strasbourg criteria (Table 1 [4]) includes two obligate and four minor criteria. Obligate criteria include chronic urticarial rash and monoclonal immunoglobulin M (IgM) or IgG gammopathy. Minor criteria include recurrent fever, neutrophilic dermal infiltrate on skin biopsy, leukocytosis or elevated C-reactive protein (CRP) and findings of abnormal bone remodeling. Using the above diagnostic criteria, one is able to establish either a definite or a probable diagnosis of Schnitzler’s syndrome. Patients satisfying both the obligate as well as two of the minor criteria are presumed to have a definite diagnosis. In the presence of IgM monoclonal gammopathy, two obligate and at least one minor criterion are needed to establish definitive diagnosis. In the presence of monoclonal gammopathy of the IgG subtype, two obligate and two minor criteria are needed to establish definitive diagnosis.
 Click to view | Table 1. Strasbourg Diagnostic Criteria |
| Case Report | ▴Top |
A 56-year-old Caucasian female, with no comorbidities, initially presented to her primary care physician’s office with complaints of severe generalized fatigue and myalgia especially in her calves and thighs. She was diagnosed with fibromyalgia and was treated supportively, without improvement of her symptoms. A year later she developed bilateral hip and knee arthralgias along with prolonged morning stiffness. She also developed intermittent fevers with temperature up to 38.9 °C. Fevers were initially nocturnal in nature, but as the disease progressed there was no diurnal difference. A non-pruritic intermittent rash which initially developed over the chest wall later progressed to a diffuse involvement. Additional symptoms of intermittent fingernail bluish discoloration and diarrhea prompted workup for Raynaud’s phenomenon and celiac disease which were negative.
Patient was referred to rheumatology and dermatology and underwent an extensive workup. Due to progressive bilateral lower extremity pain and weakness she underwent evaluation for lumbar spine stenosis with magnetic resonance imaging (MRI) of the spine. Autoimmune workup with antinuclear antibody (ANA), rheumatoid factor, perinuclear anti-neutrophil cytoplasmic antibody (p-ANCA), cytoplasmic anti-neutrophil cytoplasmic antibody (c-ANCA), SSA/SSB, SM-Ab IgG and RNP IgG testing were unrevealing. However, she was noted to have elevated inflammatory markers with CRP of 30 mg/L (< 0.5 mg/dL) and erythrocyte sedimentation rate (ESR) of 37 mm/h (0 - 25 mm/h). Evaluation for Lyme disease was negative. Additional workup revealed a monoclonal gammopathy of 0.3 g/dL.
Punch biopsy of the skin showed changes consistent with chronic urticaria (Fig. 1). The rash persisted despite treatment with antihistamines. The constellation of symptoms and elevated inflammatory markers were highly suggestive of an underlying autoimmune disorder, thus she was started on a trial of steroids. Following initial response to steroids, her symptoms recurred quickly upon tapering her steroid dose. A 6-month course of Celebrex and Plaquenil did not provide any clinical benefit and was discontinued. She underwent right hip arthroplasty due to worsening hip pain. Pathologic examination revealed portions of synovial soft tissue with acute and chronic synovitis. This was soon followed by left hip arthroplasty. Pathology showed osteocartilaginous tissue with reactive changes, reactive synovium with chronic synovitis and focal lymphoid aggregates. Pathology in neither showed changes concerning for steroid-induced osteonecrosis. Autoimmune profile was repeated, which once again revealed only elevated ESR, CRP and monoclonal gammopathy. She was started on high-dose steroids with a provisional diagnosis of adult onset Still’s disease with atypical presentation and was referred to hematology for further evaluation of her monoclonal gammopathy.
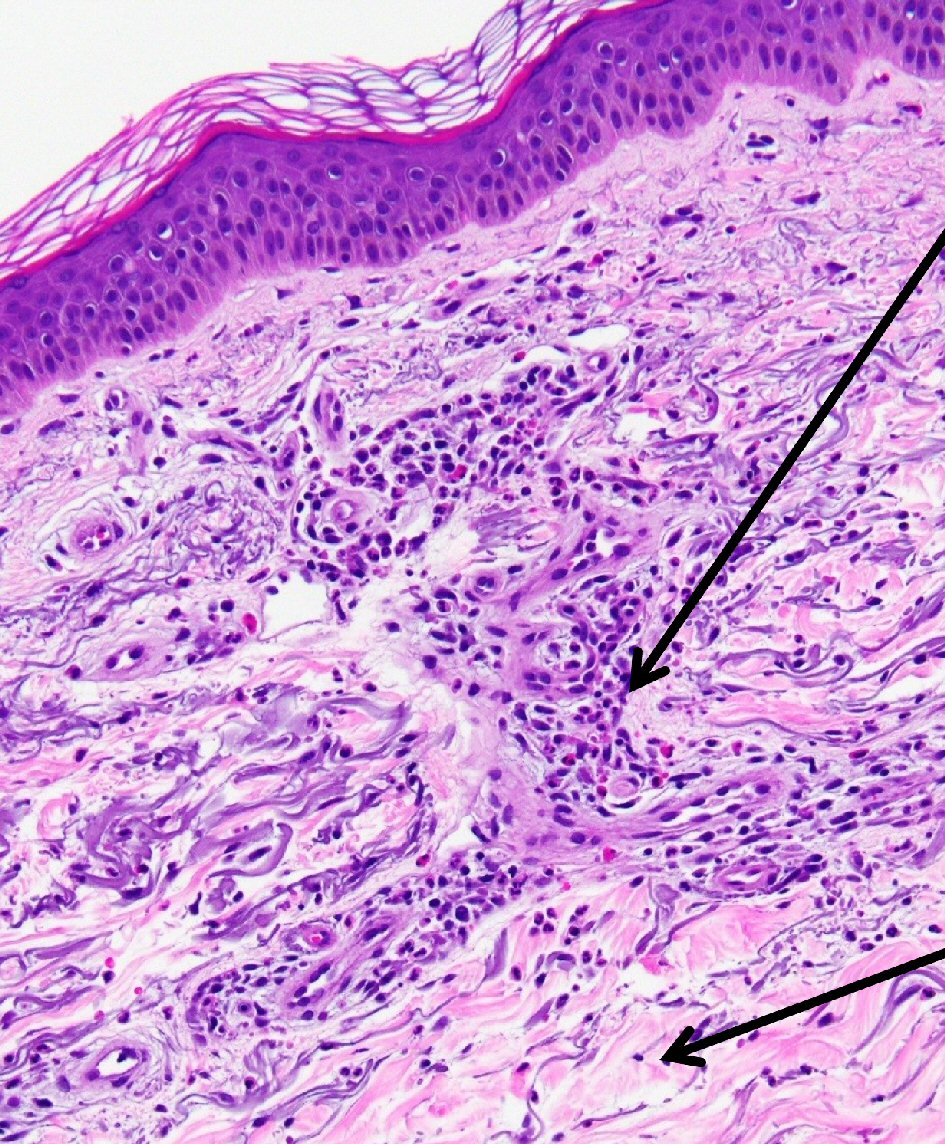 Click for large image | Figure 1. Skin punch biopsy of right forearm. Histopathologic examination revealed neutrophilic and eosinophilic infiltration of the dermis, including perivascular mixed inflammatory infiltrate suggestive of urticaria (hematoxylin and eosin (H&E), × 20). Perivascular mixed infiltrate of lymphocytes, eosinophils and neutrophils (upper arrow); surrounding sparse mixed inflammatory interstitial infiltrate with accompanying dermal edema (lower arrow). |
Evaluation by hematologist, her symptoms of fevers, arthralgias, chronic intermittent urticaria with the absence of pruritus, myalgias with a negative rheumatologic workup and presence of IgM kappa monoclonal gammopathy, raised the suspicion for Schnitzler’s syndrome. Also diagnosis of systemic mastocytosis was in the differential diagnoses. Repeat testing revealed M spike of 0.7/dL. Complete blood count showed elevated white blood cell (WBC) count of 19.9 × 103/mm3 (4.4 - 11.3 × 103/mm3), absolute neutrophil count (ANC) 17 × 103/mm3 (2.0 - 9.03 × 103/mm3), low hemoglobin at 10.6 g/dL (14 - 17.4 g/dL) with an mean corpuscular volume (MCV) of 88 fL (80 - 96 fL). Platelet count was 496 × 103/mm3 (145 - 445 × 103/mm3). She was anemic with low iron at 29 µg/dL (35 - 150 µg/dL), iron saturation of 11% (15-50%), ferritin 245 ng/mL (13 - 150 ng/mL), total iron binding capacity of 265 µg/mL (212 - 360 µg/mL). Additional anemia workup included normal B12 level at 442 pg/mL (239 - 931 pg/mL) and normal folate at 15.4 ng/mL (> 4.7 ng/mL). Lactate dehydrogenase (LDH) was normal at 212 U/L (110 - 216 U/L), and tryptase level was elevated at 19.9 ng/mL (< 11.5 ng/mL). Immunoglobulin testing revealed elevated IgE at 434 kU/L (< 214 kU/L), normal IgG at 1,040 mg/dL (700 - 1,600 mg/dL), elevated IgM at 365 mg/dL (40 - 230 mg/dL) and normal IgA at 325 mg/dL (70 - 400 mg/dL). Molecular testing was positive for MYD88, c.794T>C (p.L265P) mutation and negative for C-KIT mutation. Bone marrow biopsy showed a normocellular marrow with maturing myeloid and erythroid series without any immature forms or mast cells. A plasma cell population of 5-10% was seen without light chain restriction. Congo red stain was negative.
Based on the results of the above workup a diagnosis of Schnitzler’s syndrome was made. Patient was started on anakinra 100 mg subcutaneously daily for Schnitzler’s syndrome, and within 24 h of starting treatment she had complete resolution of fevers, arthralgia, myalgia and rash. She initiated anakinra on September 20, 2018, and continued to be on it, with no relapse of symptoms. As of to date, she has no side effects to the therapy.
| Discussion | ▴Top |
Schnitzler’s syndrome is a rare autoinflammatory disease characterized by symptoms of intermittent non-pruritic urticaria, fevers, arthralgias and monoclonal gammopathy, all mediated by overproduction of IL-1.
Our patient presented with a constellation of signs and symptoms classic for the diagnosis of Schnitzler’s syndrome, but diagnosis was delayed by almost 5 years, as happens with most cases of this syndrome. The symptoms of ongoing fatigue, fever, arthralgias, myalgias and rash are often debilitating adding to considerable morbidity. In our case it led to her leaving the work force and requiring bilateral hip replacement. The reason for such a delay in diagnosis can be attributed to rarity of the syndrome and that the presentation often mimics autoimmune or infectious disorders. This highlights the importance and need for increasing physicians’ awareness of this syndrome, not only due to potential morbidity but also because effective treatment strategies exist.
The skin rash is often the first presenting symptom [2, 5], but was not initially present in the case that we describe. The rash is clinically described as “urticarial” .On histopathological examination a neutrophilic infiltrate of the dermis is noted (Fig. 1).
Bone pain and arthralgias are common. Joint destruction has been reported, but only rarely involving the femur [2]. Herein we report bilateral hip joint destruction necessitating bilateral hip replacement. During one of the surgeries the synovial fluid was packed with WBC and the procedure had to be aborted due to concern for active joint infection. Later it was established that the nature of the finding was reactive, due to the high levels of inflammation.
The differential diagnosis of chronic urticaria, inflammation, fever and paraproteinemia is broad. Autoimmune disorders such as adult-onset Still’s disease and systemic lupus erythematosus need to be excluded. Extensive evaluation for mastocytosis, cryoglobulinemia, Waldenstrom’s macroglobulinemia, lymphoma and multiple myeloma is usually undertaken. Bone marrow examination may reveal plasmacytosis.
For patients with moderate to severe symptoms and highly elevated inflammatory markers treatment with anakinra, a recombinant IL-1 antagonist is recommended [1-3, 6]. Anakinra produces dramatic clinical response in all the patients with Schnitzler’s syndrome [7]. The responses are often durable [1, 6, 8]. Treatment failure should lead to reconsideration of the diagnosis [1, 6].
In general anakinra is well tolerated. The known side effects are grade 3-4 neutropenia and development of serious infections [6, 9].
Schnitzler’s syndrome carries a good prognosis. However, 10-20% of the cases can develop a hematologic malignancy including Waldenstrom’s macroglobulinemia, IgM multiple myeloma or non-Hodgkin lymphoma [1-3, 8, 10]. The mean duration of development of this complication is about 8 years since the onset of symptoms. Interestingly, it is noted that anakinra offsets the development of these lymphoproliferative disorders. AA amyloidosis can develop in untreated cases [2, 3, 6, 8].
Conclusions
Schnitzler’s syndrome is a rare, late-onset, acquired anti-inflammatory syndrome that should be suspected in patients with urticarial rash and monoclonal gammopathy. It should also be part of evaluation for fever of unknown origin. The presence of a constellation of clinical criteria along with monoclonal gammopathy can aid in the diagnosis of this entity. Effective treatment options include IL-1 antagonists. Prognosis is good, especially if timely diagnosis is established.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained from the patient.
Author Contributions
All the listed authors have contributed in writing the case report. Primary authors: Jyothika Mamadgi, Laila Babar, Mariya Apostolova. Expert review and opinion: Dr. Santosh Sadashiv and Dr. Rama Bhagavatula. Pathology review and slides: Dr. Kossivi Dantey.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Simon A, Asli B, Braun-Falco M, De Koning H, Fermand JP, Grattan C, Krause K, et al. Schnitzler's syndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68(5):562-568.
doi pubmed - Lipsker D. The Schnitzler syndrome. Orphanet J Rare Dis. 2010;5:38.
doi pubmed - Baresic M, Mitrovic J, Morovic Vergles J, Anic B. Different therapeutic paths (Colchicine vs. Anakinra) in two patients with Schnitzler's syndrome. Arch Rheumatol. 2016;31(4):377-380.
doi pubmed - Gusdorf L, Asli B, Barbarot S, Neel A, Masseau A, Puechal X, Gottenberg JE, et al. Schnitzler syndrome: validation and applicability of diagnostic criteria in real-life patients. Allergy. 2017;72(2):177-182.
doi pubmed - de Koning HD. Schnitzler's syndrome: lessons from 281 cases. Clin Transl Allergy. 2014;4:41.
doi pubmed - Neel A, Henry B, Barbarot S, Masseau A, Perrin F, Bernier C, Kyndt X, et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler's syndrome: a French multicenter study. Autoimmun Rev. 2014;13(10):1035-1041.
doi pubmed - Ruiz-Villaverde R, Bueno-Rodriguez A, Sanchez-Cano D. Beyond Urticaria: Schnitzler Syndrome. Balkan Med J. 2017;34(5):478-479.
doi pubmed - de Koning HD, Bodar EJ, van der Meer JW, Simon A, Schnitzler Syndrome Study G. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. 2007;37(3):137-148.
doi pubmed - Salliot C, Dougados M, Gossec L. Risk of serious infections during rituximab, abatacept and anakinra treatments for rheumatoid arthritis: meta-analyses of randomised placebo-controlled trials. Ann Rheum Dis. 2009;68(1):25-32.
doi pubmed - Gameiro A, Gouveia M, Pereira M, Tellechea O, Goncalo M. Clinical characterization and long-term follow-up of Schnitzler syndrome. Clin Exp Dermatol. 2016;41(5):461-467.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.