

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://www.thejh.org |
Original Article
Volume 11, Number 4, August 2022, pages 131-141

Experiences of a Single Center in One Hundred Ninety-Four Adult Patients With Langerhans Cell Histiocytosis
Claus Doberauera, b, d ![]() , Christoph Bornemannb, c
, Christoph Bornemannb, c
aDepartment of Internal Medicine, St. Franziskus-Hospital, Cologne, Germany
bDepartment of Internal Medicine, Protestant Clinics, Gelsenkirchen, Germany
cPractice General Medicine, Essen, Germany
dCorresponding Author: Claus Doberauer, Department of Internal Medicine, St. Franziskus-Hospital, D-50825 Koln, Germany
Manuscript submitted June 10, 2022, accepted July 18, 2022, published online August 30, 2022
Short title: Adult Patients With LCH
doi: https://doi.org/10.14740/jh1020
| Abstract | ▴Top |
Background: Langerhans cell histiocytosis (LCH) is a rare inflammatory myeloid neoplasia belonging to the group of histiocytoses. Inflammatory tissue destruction with fibrosis can result in dysfunction in any organ. Our evaluation aimed to collect information on characteristics, courses, and therapeutic options of this rare disease pattern in adult patients with conclusions on prognostic factors and follow-up management.
Methods: The medical records of 194 adult patients with histologically confirmed LCH were evaluated in this retrospective study. Patients were treated at the Protestant Clinics in Gelsenkirchen from 2000 to 2014 and St. Franziskus-Hospital in Cologne until 2020.
Results: The median age of onset was 38 years (18 to 79 years). In 65.5% of patients, only one organ was primarily involved, and in 34.5% of cases, multiple organs were involved. The skeleton, lungs, and skin were most commonly affected. In 15.5% of patients, pituitary insufficiency existed years before or at the time of diagnosis. The follow-up time of patients from the time of histologic diagnosis ranged from 6 to 408 months (median 49 months). Four patients died from sequelae of their underlying histiocytic disease. Irreversible late sequelae due to disease or therapy were detectable in 34% of patients. In 25.3% of the patients, the course of the disease could be controlled initially, but with the proviso of no smoking in case of lung involvement. Specific therapeutic measures such as surgery for solitary osteolysis, radiotherapy of osseous and cerebral manifestations, immunotherapy especially for lung and skin involvement, and chemotherapy for multisystem disease were primarily required in 74.7% of patients. As a result, 27.3% of all patients reached the nonactive stage. Of these, 26.4% had reactivation during the follow-up period. Of the remaining patients with continued active disease, 51.1% showed disease progression during follow-up.
Conclusions: Standardized diagnostics are required to capture the clinical picture. Due to the variable course, it is often sufficient to initially control with obligatory smoking cessation in case of pulmonary involvement. Follow-up examinations should be predominantly symptom-oriented with attention to possible late sequelae.
Keywords: Langerhans cell histiocytosis; Adult patients; Standardized diagnostics; Courses of disease; Treatment; Prognosis
| Introduction | ▴Top |
Histiocytic disorders are of hematopoietic origin. According to the revised classification of histiocytoses of 2016 [1], the L (Langerhans) group includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis (ICH), Erdheim-Chester disease (ECD), and overlap LCH/ECD. Immunohistochemically, the molecules CD1a, and more specifically, CD207 (Langerin) can be visualized on the surface or within Langerhans cells [2]. The earlier electron microscopic detection of Birbeck granules is no longer required for diagnosis in the presence of CD207 staining (Fig. 1). In ICH and ECD, there is no expression of CD207. The disease is associated with a various activating somatic mutations of the central signal transduction pathway mitogen-activated protein kinase (MAPK). Mutations of BRAF (especially BRAF-V600E) and MAP2K1 have been detected most frequently [3]. The mutant cells proliferate clonally and can cause tissue damage with organ dysfunction in all body organs through granuloma formation and inflammation.
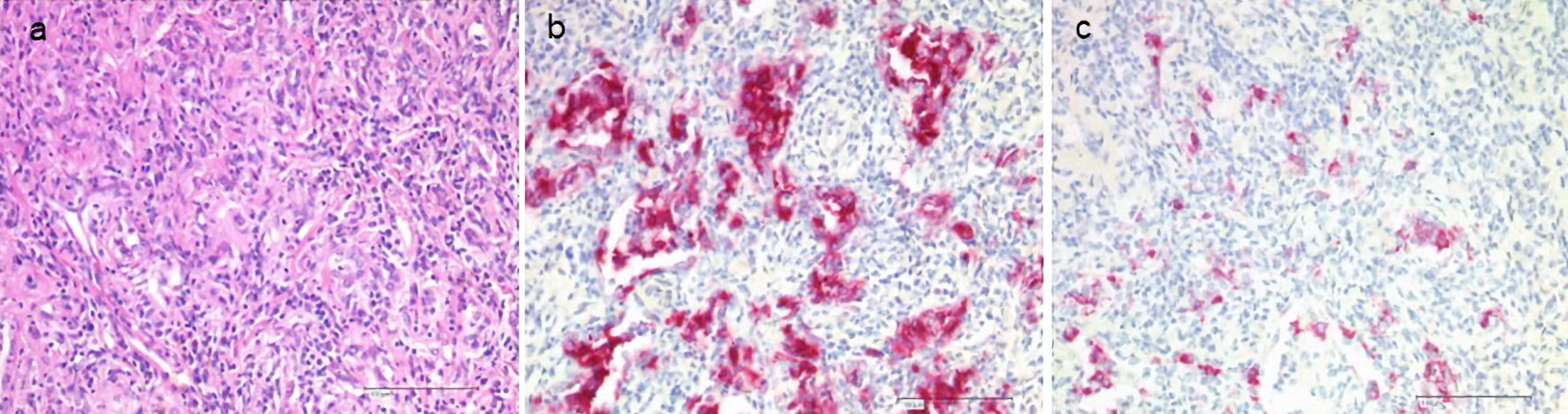 Click for large image | Figure 1. Histology of LCH in the lungs. Staining with hematoxylin and eosin (H&E, a), CD1a (b), CD207 (Langerin, c). LCH: Langerhans cell histiocytosis. |
The disease can occur at any age. The annual incidence is five to nine cases per 1 million children and adolescents younger than 15 years [4] and an estimated one to two cases per 1 million adults [5]. Familial clustering of LCH has been described [6]. Isolated lung involvement in adults correlates strictly with smoking [7]. Progression of LCH to Langerhans cell sarcoma has been documented [8].
Each organ may be affected individually (single system disease (SS)) or together with other organs (multisystem disease (MS)). Therapy depends on the pattern of involvement, disease activity, and organ dysfunction. Procedures known for many years in children cannot be transferred to adult patients without restrictions [9]. Considering new developments, especially regarding clonality and molecular genetic alterations with the possibility of targeted therapies, the existing recommendations for the diagnosis and treatment of LCH in adults have been updated [10]. With early diagnosis and therapeutic intervention, the prognosis for patients is good. The course of the disease is highly variable, with both spontaneous regressions and chronic active courses. Depending on the localization and severity of the disease as well as necessary therapeutic measures, permanent sequelae are possible.
Our study aimed to collect information on characteristics, courses, and therapeutic options of this rare disease pattern in adult patients with conclusions on prognostic factors and follow-up management.
| Materials and Methods | ▴Top |
The medical records of 194 adult patients with histologically confirmed LCH were available for the present retrospective study. These patients were treated in the Department of Internal Medicine of the Protestant Clinics Gelsenkirchen from October 2000 to June 2014 and subsequently in the Department of Internal Medicine of the St. Franziskus-Hospital in Cologne until December 2020. Patients with only probable pulmonary LCH according to the cytology of bronchioloalveolar lavage and imaging or first manifestation of LCH already in childhood or adolescence were omitted. The study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of the Medical Faculty of the Ruhr University Bochum (registration number 4266-12, 06.08.2012).
Definitions
According to the criteria used by the Histiocyte Society in randomized trials [11-13], the disease is classified into the stages of active and nonactive when all symptoms and manifestations of the disease have regressed. Active disease, in turn, is categorized as regressive (improvement of symptoms and disease signs without any new lesions), stable (persistence of symptoms and disease manifestations without any new lesions), and progressive (increase in symptoms and disease signs or appearance of new lesions). Treatment response was assessed as the response (complete regression (CR), or partial regression (PR)), intermediate response (stable course or mixed response with new lesions at one site and regressed lesions at another site, SD), and nonresponse (progressive disease (PD)). Reactivation was defined as the recurrence of disease activity regardless of site after previously CR. Late effects were considered irreversible clinical changes that developed during the disease either due to LCH or its therapy.
Statistical analysis
The statistical program XLSTAT from Addinsoft (https://www.addinsoft.com) was used to analyze the collected data. In addition to the median, the interquartile range (IQR) was reported in each case. Survival time data (overall survival and event-free survival) were analyzed using the Kaplan-Meier method with 95% confidence intervals (CIs). The log-rank test was used to compare survival curves. The significance level was set at P < 0.05.
Diagnostics
Clinical diagnostics were standardized, including anamnestic questions about smoking habits, previous and concomitant diseases, and questions about symptoms of hormonal insufficiency. During the physical examination, the entire integument was inspected, including the anogenital region, the external auditory canals, and the oral cavity, with possible involvement of particular disciplines (Fig. 2). This was also true in case of visual disturbances and neurological or psychological abnormalities. The thorax was examined by X-ray in two planes. In case of abnormalities, especially in the upper and middle lung fields, a supplementary computed tomography (CT) was performed in high-resolution technique (Fig. 3). For the question of bony manifestations, a CT in low-dose technique was performed (Fig. 4). Brain and, if necessary, spinal cord were examined by magnetic resonance imaging (MRI). This often revealed nodular thickening of the pituitary stalk with the uptake of contrast medium or failure of the neurohypophyseal signal hyperintense natively in T1 weighting in the case of LCH manifestation of the hypothalamic-pituitary region. Neck with thyroid gland and abdominal and pelvic organs were preferentially examined by sonography. Further measures such as endoscopy with biopsy collection were based on clinical symptoms, organ involvement, and the need for histologic confirmation. In case of laboratory evidence of involvement of the hematopoietic system, bone marrow aspiration was also performed. Nuclear medicine examinations such as skeletal scintigraphy and positron emission tomography (PET)/CT were used only in isolated cases. The sensitivity of skeletal scintigraphy is limited in LCH because the obligate osteolytic changes lead only to a slight increase in bone metabolic activity. Compared with conventional imaging, PET/CT can detect additional mainly osseous lesions and assess already visible changes concerning metabolic disease activity. But about lesions in the lungs and central nervous system, CT and MRI remain indispensable [14].
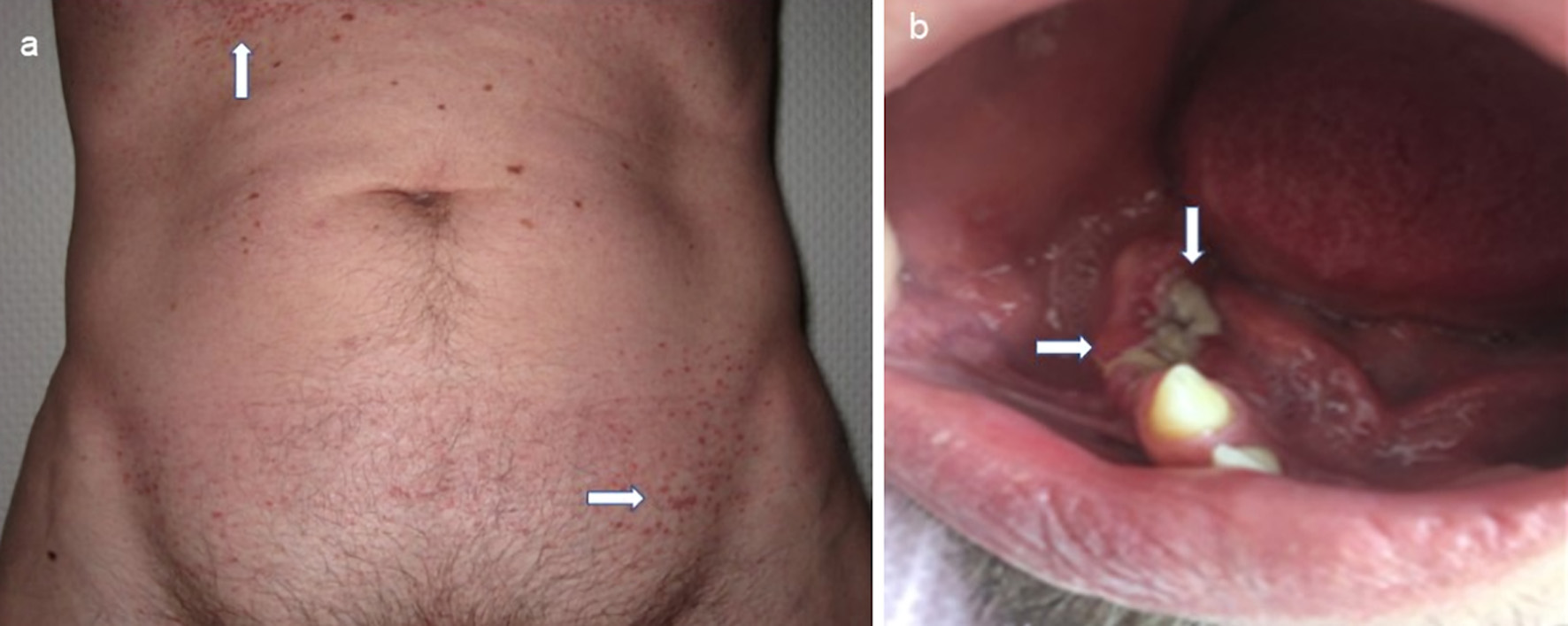 Click for large image | Figure 2. Mucocutaneous manifestations of LCH with maculopapular brownish exanthema in the area of the anterior sweat groove and in the groin region (a, indicated by arrows) and ulcerative change of the gums (b, indicated by arrows) with already tooth loss. LCH: Langerhans cell histiocytosis. |
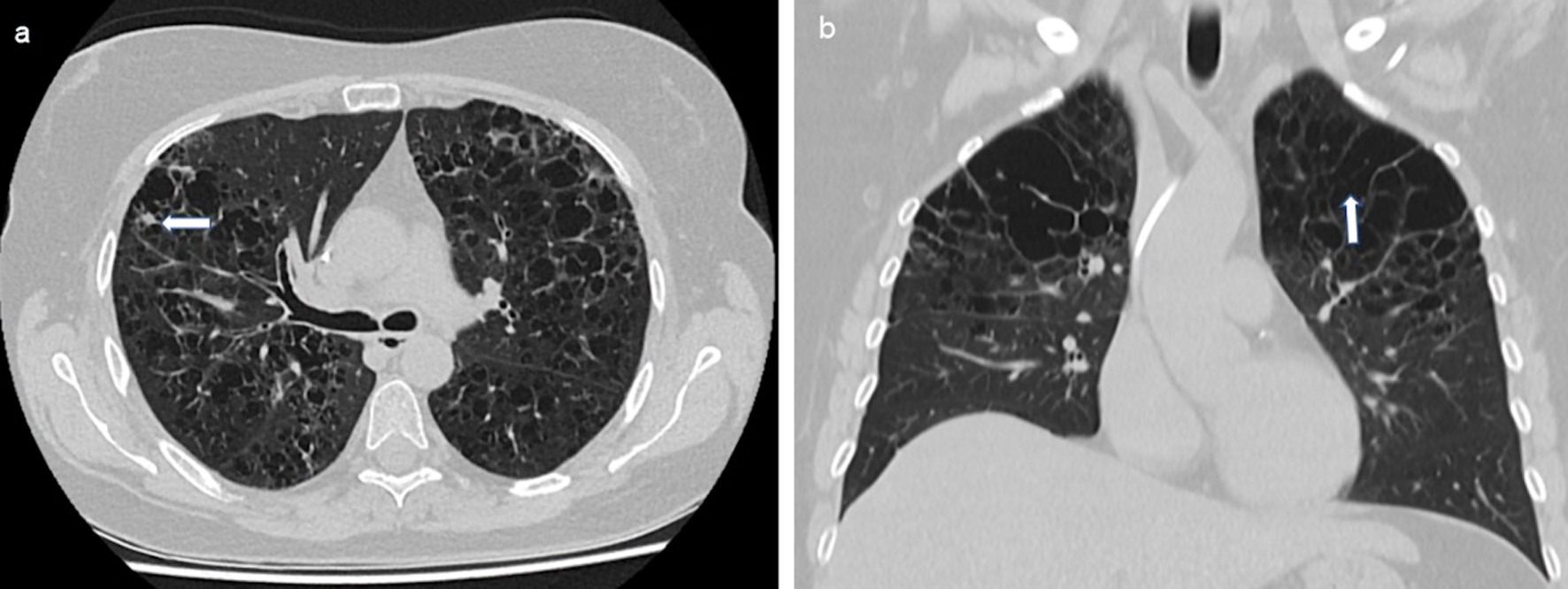 Click for large image | Figure 3. Reticulonodular compaction (a, indicated by an arrow) and cystic changes (b, indicated by an arrow) especially in the upper and middle lung fields bilaterally in LCH. CT of the thorax native, transverse sections. LCH: Langerhans cell histiocytosis; CT: computed tomography. |
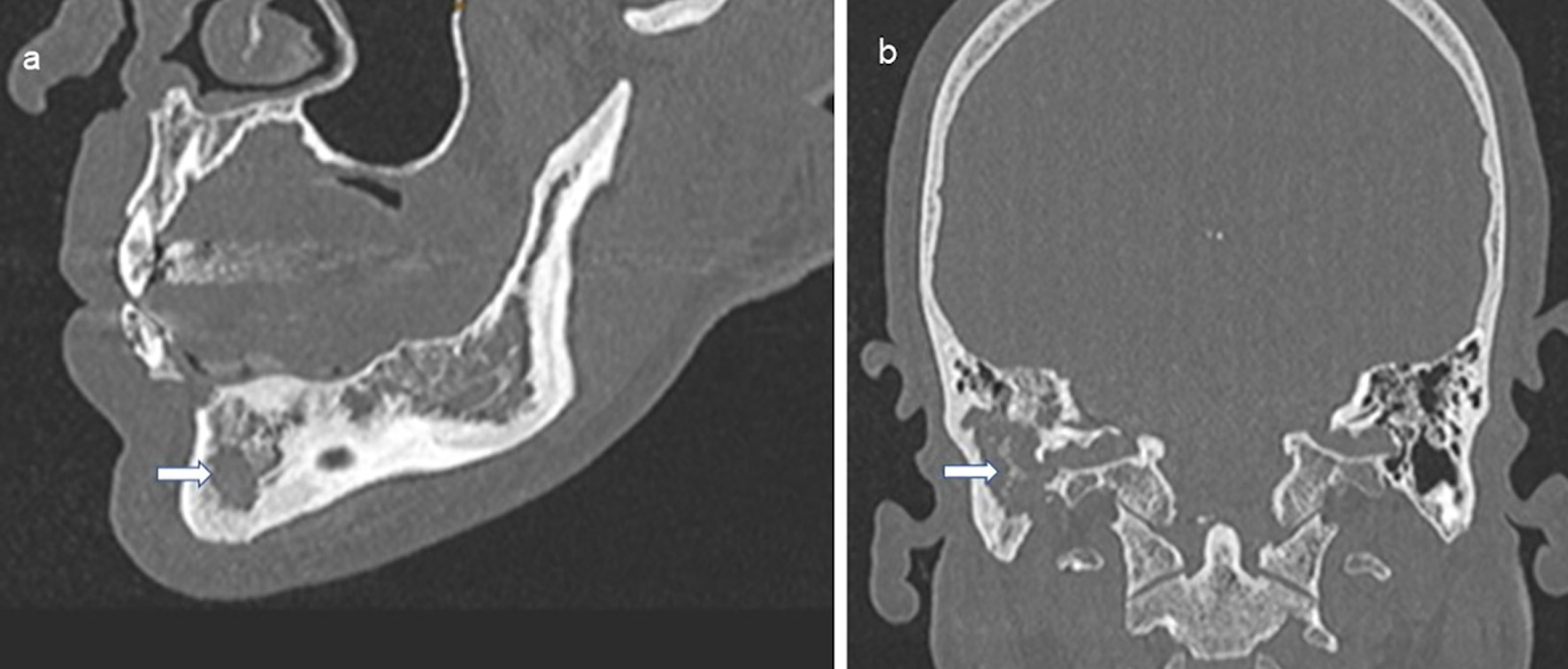 Click for large image | Figure 4. Osteolysis due to LCH in the mandible (a, indicated by an arrow) and petrous bone and temporal bone with mastoid (b, indicated by an arrow). CT of the skull native, sagittal and coronal sections. LCH: Langerhans cell histiocytosis; CT: computed tomography. |
| Results | ▴Top |
Patient characteristics
All 194 patients had histologically confirmed LCH. Taking into account the long observation period of the study, only three-quarters of the patients had undergone immunohistochemical staining with CD207 at the time of histological diagnosis. Four patients also had overlapping ECD. Additional molecular genetic analysis was performed on 15 specimens with positive evidence of a BRAF-V600E mutation in seven cases. Of the 194 patients, 89 were male (45.9%) and 105 were female (54.1%). Age at diagnosis ranged from 18 to 79 years, with a median of 38 years and IQR of 23 - 41 years. In men, the median age was 34 years (IQR 22 - 39 years), and in women, 41 years (IQR 24 - 42 years). No other cases of LCH were known in the families of our patients. Of the 194 patients, 118 were smokers (60.8%) and 76 were nonsmokers (39.2%). The observation period from histologic diagnosis was 6 to 408 months with a median of 49 months (IQR 12 - 73 months). During this period, eight patients died: two patients (LCH/ECD) aged 73 and 78 years died directly from irreversible loss of function secondary to histiocytic infiltration in brainstem and kidneys, and two patients (LCH) aged 42 and 47 years, indirectly from multisystem involvement due to sepsis with multiorgan failure under chemotherapy with neutropenia and cholangitic sepsis with liver involvement. Another four patients died independently of LCH due to suicide, chronic pancreatitis, late diabetic syndrome, and advanced atherosclerosis.
Symptoms of disease often preceded diagnosis by several months or years. This was particularly notable in 10 patients with diabetes insipidus centralis, which had been known for a median of 7.5 years (IQR 6 - 8 years), and in six patients with posterior pituitary insufficiency and partial or complete anterior pituitary insufficiency, each of unclear etiology, which had been present for a median of 2 years (IQR 1 - 5 years) (together 8.2% of patients). At the time of diagnosis, eight patients had diabetes insipidus, and another six patients had posterior and anterior pituitary insufficiency (together 7.2%). B-symptomatology consisting of fever, night sweats, and weight loss was reported by 51 patients (26.3%). Chronic non-malignant concomitant diseases were present in 45 patients (23.2%). These were autoimmune diseases in 27 patients. In 33 patients (17%), malignant disease was already detected before (18 patients), with (six patients), and after (nine patients) diagnosis of LCH. In nine cases, it was a hematologic neoplasm. Another 24 patients had solid carcinoma.
Disease manifestations
At the time of diagnosis, 127 patients (65.5%) had SS LCH uni- or multifocal, and 67 patients (34.5%) had MS LCH with involvement of preferentially two organs to disease manifestations in five organs. The localization and their distribution are listed in Table 1. The organs most frequently affected were bone, lung, and skin, singly and together with other organs. Hematopoietic dysfunction due to LCH was not present. Spleen or liver involvement was found in only one and seven patients with primary multisystem disease, respectively.
 Click to view | Table 1. Distribution of the Organs Involved at the Time of Diagnosis |
In the present evaluation, 21 adult patients had an unifocal lesion in a craniofacial bone and eight patients in the context of multifocal bone involvement. Another 27 patients had cranial bone involvement along with other organ manifestations. During follow-up, only two of the 29 patients (6.9%) with initial isolated skeletal involvement, including a craniofacial bone, developed diabetes insipidus. Among the 27 patients with the multisystem disease and a craniofacial osseous lesion, however, 12 patients already had primary signs of pituitary involvement, with two patients additionally showing cerebral focal findings or areal neurodegenerative changes. One patient (3.7%) developed the picture of a neurodegenerative disease during the course. The remaining 15 patients without primary changes in the pituitary gland or central nervous system showed no abnormalities during the course.
Therapeutic measures
In 49 patients (25.3%), a wait-and-see approach was initially possible after histologic diagnosis, with the proviso of no smoking in pulmonary manifestations. In our clientele, 39 patients had LCH limited to the lungs. Of these, 36 patients were smokers at the time of diagnosis, of whom only 20 patients self-reported cessation of nicotine abuse for at least 6 months. Among these, radiological findings in the lungs completely regressed after sustained cessation of smoking in one patient (5%) or remained stable in 16 patients (80%). In one patient with LCH in the lungs and the oral cavity, disease manifestations also completely regressed after cessation of smoking.
In contrast, 145 patients (74.7%) initially required primary specific therapy because of advanced manifestations with impending or already occurred organ dysfunction. In 42 patients (21.6%) with unifocal bone or skin findings and unclear tumors, this consisted of a diagnostically and at the same time therapeutically effective operation or excision of the disease manifestations. Two other patients (1%) had endoscopic removal of colon polyps with histologic findings of LCH with complete excision. Local recurrence occurred in three cases with surgery for solitary osteolysis of the cranial dome.
Radiotherapy was primarily used in 10 patients with skeletal involvement (seven patients with radiotherapy only, three patients with combined radiotherapy and chemotherapy). Complementary, it was performed on three patients after surgery of sclera, eyelid, or lymph nodes. Radiotherapy has been performed in 13 patients with multisystem disease because of symptomatic osseous lesions (11 patients) or central nervous system involvement with neurologic deficits (two patients). Chemotherapy was also applied in five cases. The total dose of irradiation was usually 16, 20, or 24 Gy. Overall, two local recurrences were noted after radiotherapy of an osteolysis skull calvaria with only 6 Gy and after supplemental stereotactic irradiation of the eyelid with 50 Gy.
Immunosuppressive or immunomodulatory therapy was primarily topical with a glucocorticoid for single lesions of skin and visible mucous membranes. Only in one of five cases, a long-lasting CR could be achieved. Primary systemic therapy with peroral prednisolone alone or overlapping with azathioprine or, in one case, thalidomide in cutaneous manifestations achieved SD in 17 of 19 patients with lung involvement, one CR and one PR in two of two patients with lymph node involvement, and SD in two of two patients with multifocal skin involvement.
Chemotherapy was used primarily on 44 patients (22.7%) with preferentially the cytostatics vinblastine/prednisone ± 6-mercaptopurine, cytarabine, or low-dose methotrexate and secondary on 34 patients (17.5%) with the cytostatics cladribine ± high-dose cytarabine (Fig. 5) or etoposide. During the course, 32 patients (16.5%) required multiple cytostatic treatments. The results of the most used cytostatic chemotherapy regimens are shown in Table 2.
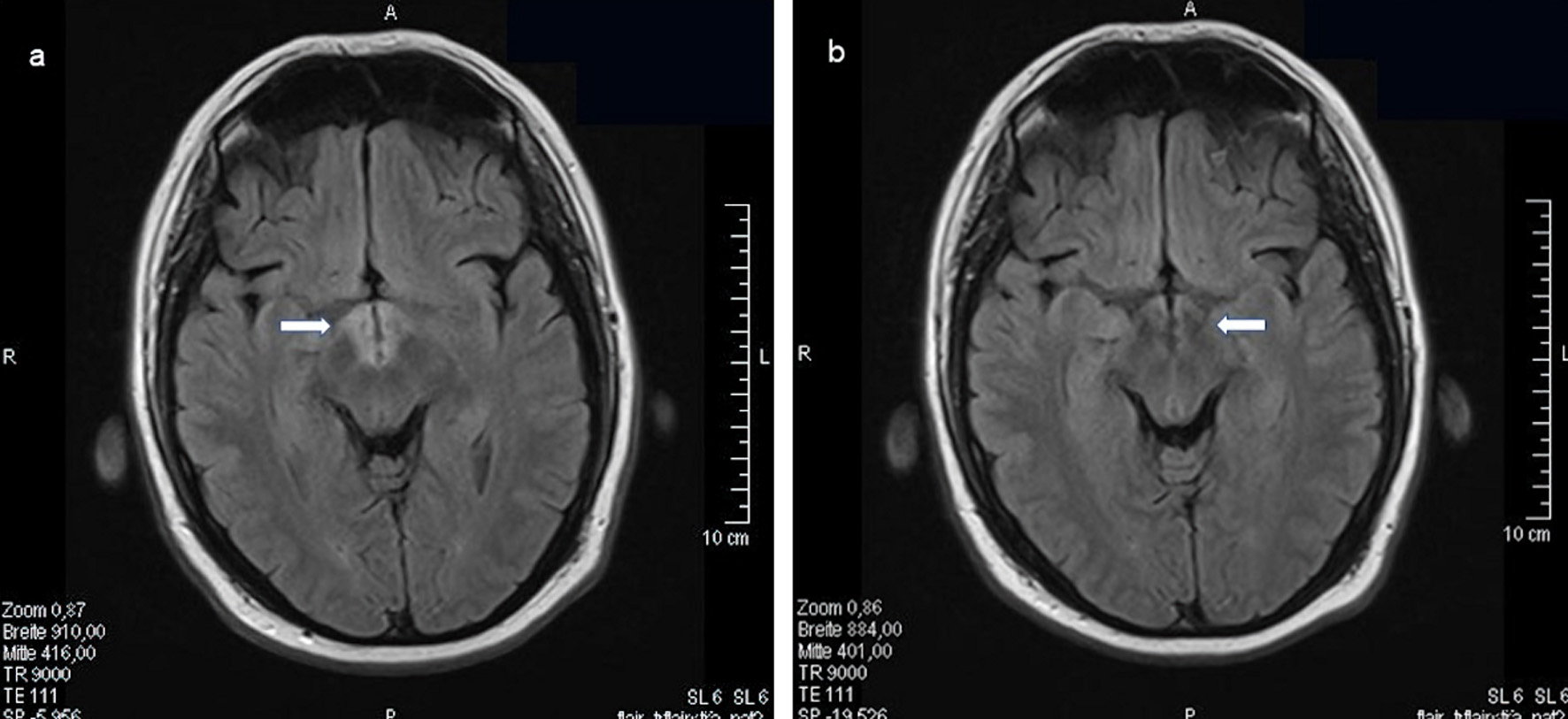 Click for large image | Figure 5. LCH infiltration of the hypothalamus bilaterally. MRI of the skull (FLAIR, transverse sections) with contrast medium before (a, indicated by an arrow) and 9 months later (b, indicated by an arrow) after cytostatic chemotherapy with high-dose cytarabine and cladribine for 3 months. LCH: Langerhans cell histiocytosis; MRI: magnetic resonance imaging. |
Click to view | Table 2. Cytostatic Chemotherapy Regimens Used |
Supportive therapy measures included bisphosphonates intravenously or perorally for multilocular skeletal involvement, psoralen perorally and UV-A light for multifocal skin involvement, and oral administration of ursodeoxycholic acid for liver involvement. Of nine patients with bisphosphonate administration alone, two patients had increasing resclerosis of osteolysis and seven patients had a stable condition.
Course of disease
After smoking cessation or specific therapy, 53 patients (27.3%) reached the disease stage nonactive. During the median follow-up period of 49 months, 14 of these patients (26.4%) experienced reactivation. In total, 11 patients (one patient with local skin involvement, 10 patients with bone involvement) showed reactivation in the originally affected organ system, but in five cases also disease manifestations in other organs. In three patients, LCH manifested again in previously uninvolved organs. In contrast, after control with smoking cessation or specific therapy, 141 patients (72.7%) remained in the active stage of disease in the following categories: active regressive in 29 of these patients (20.6%), active stable in 97 of these patients (68.8%), and active progressive in 15 of these patients (10.6%). In 72 of these patients (51%), disease activity returned or increased during the course. Chronic relapsing courses with two to four relapses requiring treatment were observed in 28 of all patients (14%).
Prognosis
Within the median follow-up period of 49 months (range: 6 - 408 months), four patients (2%) died of direct or indirect consequences of the underlying histiocytic disease. Of these, two patients had an overlap between LCH and the prognostically less favorable ECD [15]. None of the patients died from associated malignant disease. The extent of initial organ involvement in the form of SS LCH (127 patients) or MS LCH (67 patients) did not significantly affect overall survival (P = 0.398). However, a significant difference (P = 0.001) in event-free survival was observed between the disease stages nonactive (53 patients) and active (141 patients). Thus, patients without the active disease had freedom from reactivation or progression or death of a mean of 108 months (95% CI: 90.2 - 125.9 months) corresponding to a 5-year event-free survival rate of 76% (95% CI: 62.1-89.7%), and patients with active disease of a mean of 77.6 months (95% CI: 60.7 - 94.5 months) corresponding to a 5-year event-free survival rate of 43.3% (95% CI: 32.9-53.7%). But disease status did not affect overall survival (P = 0.352). In addition, liver, spleen, or central nervous system involvement without pituitary gland had no significant effect on event-free survival (P = 0.426) or overall survival (P = 0.310).
However, in 66 patients (34%), irreversible health disturbances had to be detected due to the underlying disease or the therapeutic measures taken (Table 3). These were mainly endocrine, pulmonary and neurological dysfunctions, but also disorders of the auditory and vestibular system, the dental apparatus, and the eyes. While hormonal dysfunctions, in particular, could be compensated by substitution therapy, destruction of lung parenchyma, osteolytic destruction of petrous bone and mastoid, ocular involvement, and neurodegenerative changes were associated with invalidation or significant reduction of the patients’ quality of life. One patient with primary multisystem disease and implementation of three chemotherapies developed blood count changes during the course as a manifestation of chronic myelomonocytic leukemia or myelodysplastic syndrome. Liver cirrhosis developed in one patient.
Click to view | Table 3. Long-Term Effects After Disease and Therapy |
| Discussion | ▴Top |
Experience with LCH in adult patients is limited. Currently, a group of experts from multiple subspecialities has summarized the state of the art regarding diagnosis and treatment of LCH in adult patients and formulated key recommendations [10]. The first overview of LCH in adults was provided by an evaluation of the International Histiocyte Society Registry of 274 adult patients from 13 countries [16]. It was already noticeable that the symptoms of the disease, which varied according to organ involvement, usually preceded the histological confirmation of the diagnosis by months or even years. This concerned, in particular endocrine disorders of the pituitary gland with diabetes insipidus present in 29.6% of cases. The late assignment of such an endocrine disorder to the underlying disease of LCH is undoubtedly due to the problematic fine-tissue diagnostic confirmation in this brain region. In our study, 8.2% of the patients showed hormonal signs of insufficiency of the neuro- and/or adenohypophysis in the run-up and again 7.2% at the time of histological confirmation of LCH. The pituitary region may be a preferred site of LCH due to the robust vascularization with a dense plexus of capillaries and the partial absence of a blood-brain barrier [17].
Among our patients, the proportion of smokers was very high at 60.8%. In the case of isolated lung involvement, 92.3% and in the case of lung involvement as part of a multisystem disease, 87.5% of patients reported nicotine abuse. The registry study showed 77% and 53%, respectively. Since only 10.1% of nonsmokers had lung pulmonary involvement, and radiologically there were single CRs and multiple pulmonary disease arrests in several of our patients with smoking cessation, the assumption of a pathophysiological relationship between smoking and pulmonary manifestations of LCH is supported. These findings correlate with the radiological follow-up results according to smoking status of patients with newly diagnosed LCH of the lungs without immunosuppressive treatment [18]. Smoking cessation therefore represents a fundamental behavioral measure in pulmonary involvement.
Lesions of LCH predominantly show an inflammatory infiltrate with a significant population of T cells and evidence of inflammatory cytokines. Since autoimmune diseases are also associated with an inflammatory process, it is noteworthy that 13.9% of patients in our cohort had concomitant autoimmune disease. Compared to the prevalence of an autoimmune disease of 5-8% in the German population, autoimmune diseases were clearly more prevalent in our patients.
Early on, an increased correlation with malignant disease was noted, especially in children but also in adults with LCH [19-21]. A genetic predisposition or the development of a malignancy secondary to therapy of LCH was suspected. Most frequently, malignancies were detected concurrently with or after diagnosis of LCH. Malignant lymphomas and leukemias were significantly more common than solid tumors, particularly in children. In the international registry study, the rate of concomitant malignancies at diagnosis was 6.2% with hematologic and solid neoplasms in equal proportions. In our study, 12.4% of patients had a malignancy at the time of diagnosis. Overall, as many as 17% of patients with the malignant disease were already diagnosed before LCH (18 patients), at the same time (six patients), or later (nine patients). In 27.3% of cases, this was a hematologic neoplasm and in 72.7% of cases, it was a solid carcinoma. A recent evaluation of 132 adult patients with LCH showed in 32% of patients a solid tumor (74%) or hemoblastosis (26%). The solid tumors usually preceded the diagnosis of LCH [22]. In this study, the median age of LCH patients was relatively high at 54 years compared with 38 years in our patients, which may help explain the higher number of previous malignancies. Mutations in the MAPK signal transduction pathway, which are found in various malignancies as well as histiocytic diseases, represent a possible explanation for the increased association of LCH with malignancies. This has already been demonstrated in some patients with histiocytic diseases from the L (Langerhans) group and additional hematologic neoplasia [23]. A consequence of therapy appears to be of secondary importance in adult patients with lower numbers of subsequently diagnosed malignancies. In our patients, there was only one patient with the multisystem disease developed differential chronic myelomonocytic leukemia or myelodysplastic syndrome in the course after three different chemotherapy regimens and irradiation in the jaw region.
Of the 274 patients in the Histiocyte Society International Registry (median age at diagnosis 35 years, 52% men, 46% women), 31.4% had SS LCH, and 68.6% had MS LCH. In contrast, our 194 patients (median age 38 years, men 45.9%, women 54.1%) were more likely to have SS LCH (65.5%) and were less likely to have MS LCH (34.5%) at diagnosis. Discrepancies were also evident in the distribution of organs involved. In our patient cohort, the skeleton (57.7%) was most frequently involved, followed by involvement of the lungs (40.7%) and skin (17.5%). In the organ distribution of patients included in the registry, lungs (58.4%) and skeleton (57.3%) were leading, followed by skin involvement (36.9%). The interval between diagnosis and last follow-up was shorter, with a median of 28 months, compared with a median of 49 months in our study. During the follow-up period, 15 of 274 patients (5.5%) in the registry study and eight of 194 patients (4.1%) in our study died. Our results are comparable to a retrospective evaluation by a single center in the United States of 186 adult patients (median age 43 years, 46% men, 54% women), 62% of whom had primary SS LCH, and 38% had MS LCH [24]. Among these, the most commonly affected organs were lung (59%), skeleton (37%), skin (21%), and central nervous system, including pituitary (16%). The median follow-up time was 23 months. A total of 21 patients (11.3%) died, 10 of whom died due to LCH. In contrast, another large retrospective study from a single center in China of 266 adults (median age 32 years, men 66.5%, women 33.5%) with LCH showed, as in the registry study, a high proportion of MS LCH at 68.4% with only 31.6% SS LCH [25]. The most commonly involved organs were skeletal (68.4%), lung (48.5%), pituitary (44%), and skin (18.8%). The median follow-up time was 38 months in patients with SS-LCH with only unifocal bone involvement, and 43 months in patients with multifocal bone involvement SS-m LCH or MS LCH. In both groups, two and 17 patients (a combined 7.1%) died, respectively.
In children, organs at risk for the course of the disease are considered to involve the bone marrow, liver, and spleen [11, 12, 26]. In the Chinese study, 39 patients had liver involvement and 11 patients had spleen involvement at the time of diagnosis. These had significantly worse event-free survival than patients without such organ involvement, and in the case of age older than 50 years at diagnosis, also worse overall survival. We could not replicate this association in our patient population, with only seven patients with liver involvement and one patient with splenic involvement. In the international registry study, liver or lung involvement were also without effect on overall survival. Therefore, organ risk in adult patients with LCH remains unanswered.
Furthermore, in our clientele of adult patients with LCH, the significance of osteolytic changes of the craniofacial bones concerning an increased risk of the development of diabetes insipidus or neurodegenerative changes of the central nervous system [17, 27], which is relevant for therapy in children, could not be reproduced. A separate evaluation of 29 patients with isolated bone involvement, including craniofacial bone, showed the occurrence of diabetes insipidus in only two patients (6.9%) within the median follow-up period of 49 months. Thus, craniofacial bone involvement alone in adults with LCH does not represent a risk constellation for the further course of the disease and therefore does not warrant additional systemic therapy. In contrast, 12 of 27 patients (44.4%) with craniofacial bone involvement in multisystem disease already had primary evidence of partial or complete hypopituitarism and, in two of these cases, also of central nervous system involvement. However, only one patient developed the picture of neurodegenerative disease during the course. Nevertheless, the high coincidence of bone involvement, including the skull and central nervous system involvement with the pituitary, underscores the need for a primary MRI of the skull and, if necessary, hormonal studies as part of the standardized diagnostic workup.
The course of LCH is characterized by spontaneous regression, especially in the skin, and by long-lasting disease arrest, especially in the bones after biopsy and in the lungs after smoking cessation. In the absence of impending complications or organ dysfunction, the spontaneous course of the disease can be awaited after histological diagnosis, including the recommendation to refrain from smoking. This applied to 25.3% of patients in our study. In 74.7% of cases, specific therapy measures had to be initiated first. As a result, 35.2% of specifically treated patients had disease converted to a nonactive stage. In 64.8% of patients, the disease remained active after therapy, but mainly in the active stable and active regressive categories.
Of a combined 53 patients who no longer showed active disease after smoking cessation or specific therapy, 14 patients (26.4%) developed reactivation during the follow-up period, preferentially in the same organ as at baseline. Of the 141 patients with continued active disease, 72 patients (51.1%) continued or had disease progression during follow-up; and 14.4% of all patients had a chronic relapsing course with up to four relapses. Surgery was effective in the long term, especially for solitary osteolysis. In the case of radiotherapy, total radiation doses of preferably 16 to 24 Gy resulted in CR of osseous and cerebral manifestations. Immunosuppressive therapy often stabilizes the disease in the lungs. Topical therapy in the skin area usually provided only temporary improvement. Chemotherapy was required primarily or secondarily in a total of 40.2% of all patients. The cytostatic drugs cladribine and cytarabine alone or in combination, as well as the combination vinblastine/prednisone/6-mercaptopurine, showed the strongest efficacy with an overall treatment response of 41-76%. These were predominantly PRs over a median of 21 - 39 months. In the international registry study, 47.6% of patients initially received cytostatic chemotherapy [16]. This consisted predominantly of vinblastine in combination with prednisone. No data were reported on treatment response. In the two large single-center trials in the United States and China, 42% and 65% of patients received cytostatic treatment, with cladribine- and cytarabine-based chemotherapies producing the best results [24, 25]. Promising for the future is the use of BRAF and MEK inhibitors in patients with corresponding mutations in the MAPK/ERK signal transduction pathway. Impressive study results are already available in patients with histiocytic diseases [28-30]. However, a prerequisite is molecular pathological analysis of the tissue samples. According to the international consensus recommendations, this analysis should already be carried out primarily. Otherwise, it should be performed at the latest in the case of therapy-refractory or recurrent disease progression.
The prognosis of patients is good if diagnosed early. This applies to both the nonactive and active stage of the disease. However, event-free survival is significantly longer in nonactive disease than in active diseases. Irreversible late effects due to disease or therapy represent a significant problem. In our clientele, 34% of patients had primarily endocrine, pulmonary, or neurological sequelae. Control examinations are recommended to avoid these and treat active disease relapses in time. In the case of inactive disease, symptom-oriented monitoring can usually be performed after two inconspicuous examinations at intervals of 6 months. In the case of active disease, the control intervals depend on the dynamics of the disease with possible organ dysfunction, so control intervals of 3 to 6 months are reasonable.
The present study has some limitations. It is a retrospective single-center study with standardized diagnostics but without therapy randomization. Of the tissue samples, only a tiny proportion were examined by molecular pathology. PET/CT for diagnosis, response assessment and monitoring were performed only in single cases. In the future, prospective studies should consider current developments in diagnostics and therapy according to international consensus recommendations.
In conclusion, our study shows that standardized diagnostics are required to capture the clinical picture. Due to the variable course, it is often sufficient to control initially only with an obligatory smoking cessation in case of lung involvement. Therapeutic measures depend on organ involvement, disease activity, and organ dysfunction. Follow-up examinations should be predominantly symptom-oriented with attention to possible late sequelae.
Acknowledgments
The authors would like to thank Ozlem Krischek and Ulf Laufer for the evaluation of the radiological and magnetic resonance imaging findings. Further thanks are due to Andrea Tannapfel for histopathological support.
Financial Disclosure
There was no specific financial disclosure or funding source for this study.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Author Contributions
CD contributed to study conception, data analysis with interpretation and wrote the manuscript; CB collected and analyzed data; both authors revised the paper and approved the submitted version.
Data Availability
Data supporting the findings of this study are available from the corresponding author on reasonable request.
Abbreviations
CI: confidence interval; CR: complete regression; CT: computed tomography; ECD: Erdheim-Chester disease; ICH: indeterminate cell histiocytosis; IQR: interquartile range; LCH: Langerhans cell histiocytosis; MAPK: mitogen-activated protein kinase; MRI: magnetic resonance imaging; MS: multisystem disease; ORR: overall remission rate; PET: positron emission tomography; PD: progressive disease; PR: partial regression; SD: stable course or mixed response; SS: single system disease
| References | ▴Top |
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672-2681.
doi pubmed - Collin M, Bigley V, McClain KL, Allen CE. Cell(s) of origin of Langerhans cell Histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):825-838.
doi pubmed - Rollins BJ. Genomic alterations in Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):839-851.
doi pubmed - Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer. 2008;51(1):71-75.
doi pubmed - Baumgartner I, von Hochstetter A, Baumert B, Luetolf U, Follath F. Langerhans'-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28(1):9-14.
doi - Arico M, Nichols K, Whitlock JA, Arceci R, Haupt R, Mittler U, Kuhne T, et al. Familial clustering of Langerhans cell histiocytosis. Br J Haematol. 1999;107(4):883-888.
doi pubmed - Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans'-cell histiocytosis in adults. N Engl J Med. 2002;346(7):484-490.
doi pubmed - Lee JS, Ko GH, Kim HC, Jang IS, Jeon KN, Lee JH. Langerhans cell sarcoma arising from Langerhans cell histiocytosis: a case report. J Korean Med Sci. 2006;21(3):577-580.
doi pubmed - Girschikofsky M, Arico M, Castillo D, Chu A, Doberauer C, Fichter J, Haroche J, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis. 2013;8:72.
doi pubmed - Goyal G, Tazi A, Go RS, Rech KL, Picarsic JL, Vassallo R, Young JR, et al. International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood. 2022;139(17):2601-2621.
doi pubmed - Gadner H, Grois N, Arico M, Broadbent V, Ceci A, Jakobson A, Komp D, et al. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138(5):728-734.
doi pubmed - Gadner H, Grois N, Potschger U, Minkov M, Arico M, Braier J, Broadbent V, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood. 2008;111(5):2556-2562.
doi pubmed - Gadner H, Minkov M, Grois N, Potschger U, Thiem E, Arico M, Astigarraga I, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121(25):5006-5014.
doi pubmed - Ferrell J, Sharp S, Kumar A, Jordan M, Picarsic J, Nelson A. Discrepancies between F-18-FDG PET/CT findings and conventional imaging in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2021;68(4):e28891.
doi pubmed - Arnaud L, Hervier B, Neel A, Hamidou MA, Kahn JE, Wechsler B, Perez-Pastor G, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011;117(10):2778-2782.
doi pubmed - Arico M, Girschikofsky M, Genereau T, Klersy C, McClain K, Grois N, Emile JF, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003;39(16):2341-2348.
doi - Grois N, Potschger U, Prosch H, Minkov M, Arico M, Braier J, Henter JI, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer. 2006;46(2):228-233.
doi pubmed - Tazi A, de Margerie C, Naccache JM, Fry S, Dominique S, Jouneau S, Lorillon G, et al. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis. 2015;10:30.
doi pubmed - Egeler RM, Neglia JP, Puccetti DM, Brennan CA, Nesbit ME. Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer. 1993;71(3):865-873.
doi - Egeler RM, Neglia JP, Arico M, Favara BE, Heitger A, Nesbit ME. Acute leukemia in association with Langerhans cell histiocytosis. Med Pediatr Oncol. 1994;23(2):81-85.
doi pubmed - Egeler RM, Neglia JP, Arico M, Favara BE, Heitger A, Nesbit ME, Nicholson HS. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am. 1998;12(2):369-378.
doi - Ma J, Laird JH, Chau KW, Chelius MR, Lok BH, Yahalom J. Langerhans cell histiocytosis in adults is associated with a high prevalence of hematologic and solid malignancies. Cancer Med. 2019;8(1):58-66.
doi pubmed - Kemps PG, Hebeda KM, Pals ST, Verdijk RM, Lam KH, Bruggink AH, de Lil HS, et al. Spectrum of histiocytic neoplasms associated with diverse haematological malignancies bearing the same oncogenic mutation. J Pathol Clin Res. 2021;7(1):10-26.
doi pubmed - Goyal G, Hu M, Young JR, Vassallo R, Ryu JH, Bennani NN, Shah MV, et al. Adult Langerhans cell histiocytosis: a contemporary single-institution series of 186 patients. J Clin Oncol. 2019;37(15_suppl):7018.
doi - Cao XX, Duan MH, Zhao AL, Cai H, Chen J, Gao XM, Liu T, et al. Treatment outcomes and prognostic factors of patients with adult Langerhans cell histiocytosis. Am J Hematol. 2022;97(2):203-208.
doi pubmed - Minkov M, Grois N, Heitger A, Potschger U, Westermeier T, Gadner H, DAL-HX Study Group. Response to initial treatment of multisystem Langerhans cell histiocytosis: an important prognostic indicator. Med Pediatr Oncol. 2002;39(6):581-585.
doi pubmed - Haupt R, Nanduri V, Calevo MG, Bernstrand C, Braier JL, Broadbent V, Rey G, et al. Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society-Late Effects Study Group. Pediatr Blood Cancer. 2004;42(5):438-444.
doi pubmed - Bhatia A, Ulaner G, Rampal R, Hyman DM, Abdel-Wahab O, Durham BH, Dogan A, et al. Single-agent dabrafenib for BRAF(V600E)-mutated histiocytosis. Haematologica. 2018;103(4):e177-e180.
doi pubmed - Diamond EL, Durham BH, Ulaner GA, Drill E, Buthorn J, Ki M, Bitner L, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567(7749):521-524.
doi pubmed - Hazim AZ, Ruan GJ, Ravindran A, Abeykoon JP, Scheckel C, Vassallo R, Ryu JH, et al. Efficacy of BRAF-inhibitor therapy in BRAF(V600E) -mutated adult Langerhans cell histiocytosis. Oncologist. 2020;25(12):1001-1004.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.