









| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 7, Number 4, December 2018, pages 158-162
Vasculitis in Myelodysplastic Syndrome and Chronic Myelomonocytic Leukemia: A Report of Two Cases
Justin Jacobsea, Yvo W.J. Sijpkensa, Jan W. van ‘t Wouta, Elke E.M. Petersb, L. Tom Vlasvelda, c
aDepartment of Internal Medicine, HMC Bronovo, The Hague, Netherlands
bDepartment of Pathology, HMC Bronovo, The Hague, Netherlands
cCorresponding Author: L.T. Vlasveld, HMC Bronovo, Bronovolaan 5, 2597 AX, The Hague, Netherlands
Manuscript submitted September 7, 2018, accepted October 18, 2018
Short title: Vasculitis in Myelodysplastic Syndrome
doi: https://doi.org/10.14740/jh461w
| Abstract | ▴Top |
There is a clear association between myelodysplastic syndrome (MDS)/chronic myelomonocytic leukemia (CMML) and autoimmune manifestations such as vasculitis. It is not clear if autoimmune manifestations in myelodysplastic syndrome are a cause or consequence. We describe two patients with polyarteritis nodosa and large vessel vasculitis, as presenting symptom of a myelodysplastic syndrome with excess blasts type 2 and chronic myelomonocytic leukemia respectively. Immunosuppressive treatment resulted in amelioration of the vasculitis with improvement of the myelodysplastic features in the first patient and rapid evolution to acute myeloid leukemia in the other patient. The association between MDS/CMML and autoimmune manifestations, such as vasculitis, emphasizes the role of autoimmunity in the clinical features and even pathogenesis of MDS/CMML.
Keywords: Autoimmune manifestation; Chronic myelomonocytic leukemia; Myelodysplastic syndrome with excess blasts; Vasculitis; Immunosuppression
| Introduction | ▴Top |
Myelodysplastic syndrome (MDS) is a heterogeneous group of clonal neoplasms, characterized by dysplastic features in hematopoietic cells and peripheral cytopenia with a variable risk for development of acute myeloid leukemia (AML) [1]. Since the first description in 1938, growing insight into the pathophysiology has led to regular revision of the classification [1, 2]. Chronic myelomonocytic leukemia (CMML), initially categorized within MDS, is currently classified as myelodysplastic/myeloproliferative neoplasm [1, 3]. There is a correlation between MDS/CMML and the presence of a broad spectrum of autoimmune manifestations (AIM) [4-9]. Patients with autoimmune diseases have an increased risk for the development of MDS with an odds ratio of 1.5 to 3.5, and clinically apparent AIM occur in 5-35% of the MDS, while up to 60% of MDS patients may have serological signs of autoimmunity [8, 9]. The incidence of AIM is likely to be irrespective of subtype of MDS and prognostic factors, such as cytogenetic abnormalities [8, 9]. The survival of MDS patients with and without AIM is usually reported to be similar, although a better survival and less risk for the development to acute myeloid leukemia (AML) is found in occasional studies [8-10]. Within the AIM vasculitis predominate with an incidence from 5 to 60% [5, 9-11]. In MDS, cutaneous small vessel/leukocytoclastic vasculitis is most frequent [9, 12]. Systemic vasculitis of medium-sized vessels such as polyarteritis nodosa (PAN) and large-sized vessels such as giant-cell arteritis and Takayashu’s disease are less frequent and likely to be associated with CMML [7, 8, 12]. We present two patients with PAN and giant cell arteritis, as presenting symptom of MDS with excess blasts type 2 (MDS-EB-2) and CMML respectively. Immunosuppressive treatment resulted in amelioration of the vasculitis. While there was improvement of the myelodysplastic features in the first patient, there was a rapid evolution to AML in the other patient. These cases fuel discussion of the immune system balance in MDS.
| Case Reports | ▴Top |
Case 1
A 77-year-old female without relevant medical history was admitted because of multiple reddish-purple nodules on the lower legs (Fig. 1) and fever up to 39.3 °C for 1 day. Blood analysis revealed pancytopenia with elevated inflammatory parameters, normal renal and liver function tests, and diffuse hypergammaglobulinemia (Table 1). An infectious focus was not found. Histological examination of a skin nodule showed a vasculitis of small and medium-sized arteries, with moderate eosinophilia (Fig. 2).
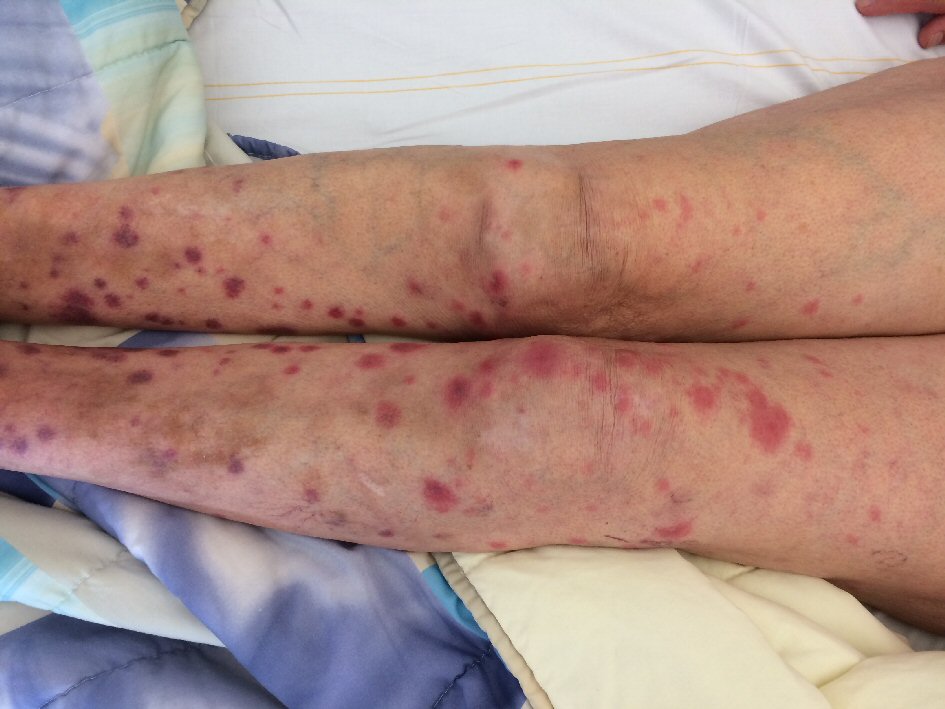 Click for large image | Figure 1. Multiple reddish-purple nodules on the legs of patient A. |
 Click to view | Table 1. Laboratory Results of Patients A and B at Presentation |
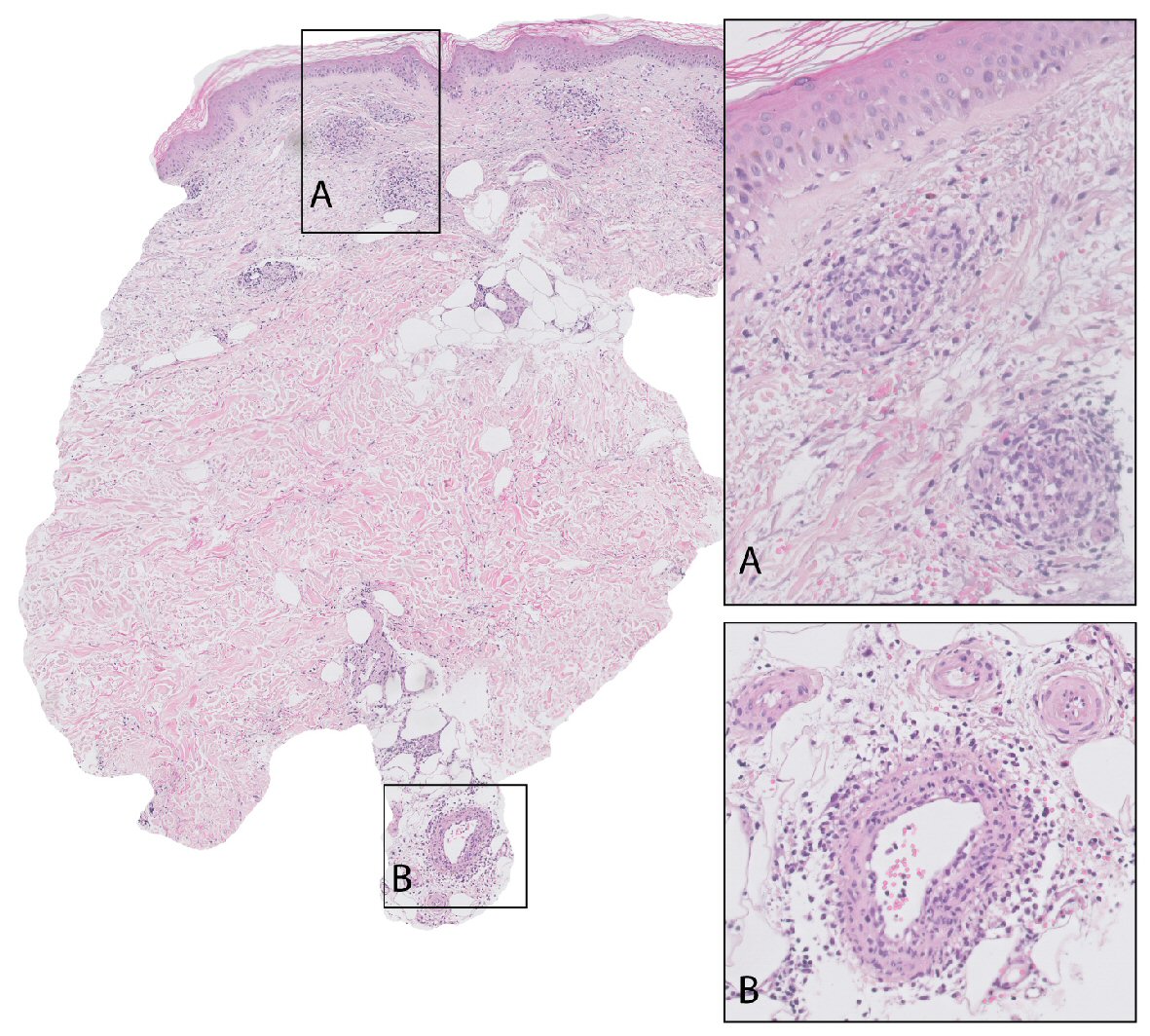 Click for large image | Figure 2. Skin biopsy taken from the left lower leg of patient A. It shows epidermis, dermis and a medium-sized vessel in the upper subcutis. Small vessels (detail in box A) as well as medium-sized vessels (detail in box B) throughout the depth of the biopsy are circumferentially infiltrated by inflammatory cells, mainly lymphocytes. Endothelial cells are swollen and infiltrated by lymphocytes, narrowing the vessel lumen. There are no fibrinous thrombi or necrotic vessels. Affected vessels are surrounded by nuclear dust, erythrocytes and some eosinophils. |
An extensive panel of autoimmune antibodies and serologic tests for hepatitis B and C were negative. Computed tomography (CT) thorax and abdomen showed hepatomegaly without splenomegaly or lymphadenopathy. Bone marrow examination revealed hypercellularity with dysplasia in erythropoiesis and megakaryopoiesis and 10% myeloid blasts. Cytogenetic testing revealed an isochromosome in the short arm of chromosome X (X, i(X)), with undetermined prognostic significance. Prednisolone treatment was initiated for MDS-EB-2 associated cutaneous PAN.
Due to recurrent episodes of non-infectious fever and PAN exacerbations in the subsequent year, it was impossible to taper the prednisone dosage. Approximately 1 year after initial diagnosis and during treatment with 30 mg prednisone daily, the patient was admitted because of new urticaria-like, non-hardened, red plaques, some of which had a target lesion appearance. In contrast, the PAN of the lower legs showed signs of regression. Histological examination of a new lesion showed vasculitis of the superficial vessels, similar as to what was observed before. The biopsy was shallow, not reaching the junction with the subcutis where medium-sized vessels are situated, therefore resulting in a non-diagnostic biopsy for diagnosis of PAN.
Since it was impossible to decrease the prednisone dosage azathioprine 50 mg once daily was added, after which the prednisone dosage could be tapered. Eventually, the signs of the vasculitis subsided. During the immunosuppressive treatment the Hb increased from 9.3 to 12 g/dL (normal range (NR): 12.8 - 16.8), the leucocytes from 1.4 to 4.5 ×106/mL (NR: 4.0 - 10.0) and the thrombocytes from 66 to 132 × 106/mL (NR: 150 - 400). In the bone marrow the percentage blasts had decreased from 10% to 4% with less prominent dysplastic features. Cytogenetic studies still revealed the presence of X, i(X).
Case 2
A 68-year-old male with adult onset diabetes mellitus was evaluated at the outpatient clinic because of muscle weakness in the legs, headache, night-sweats, 5 kg weight loss and fever up to 39 °C. The temporal arteries, bilateral blood pressure and peripheral pulsations were normal. He had no lymphadenopathy or hepatosplenomegaly. Laboratory examination (Table 1) revealed an elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), macrocytic anemia and monocytosis (4.3 × 106/mL), while 13 months earlier the blood counts were normal including a normal MCV and no monocytosis. The biochemical profile was unremarkable including a normal lactate dehydrogenase and immunoelectrophoresis.
CT of the thorax and abdomen was normal. In the workup of fever of unknown origin with an elevated ESR and CRP a positron emission tomography (PET) scan was performed, showing an increased up-take of 18F-fludeoxyglucose in the ascending part and arch of the aorta (Fig. 3); compatible with large vessel vasculitis (Takayashu’s disease) in the context of an underlying myelodysplastic/myeloproliferative neoplasm. At that time bone marrow biopsy was declined, but in view of the peripheral monocyte count of 4.3 × 106/ml, being 36% of the leucocyte count, CMML was the presumed working diagnosis.
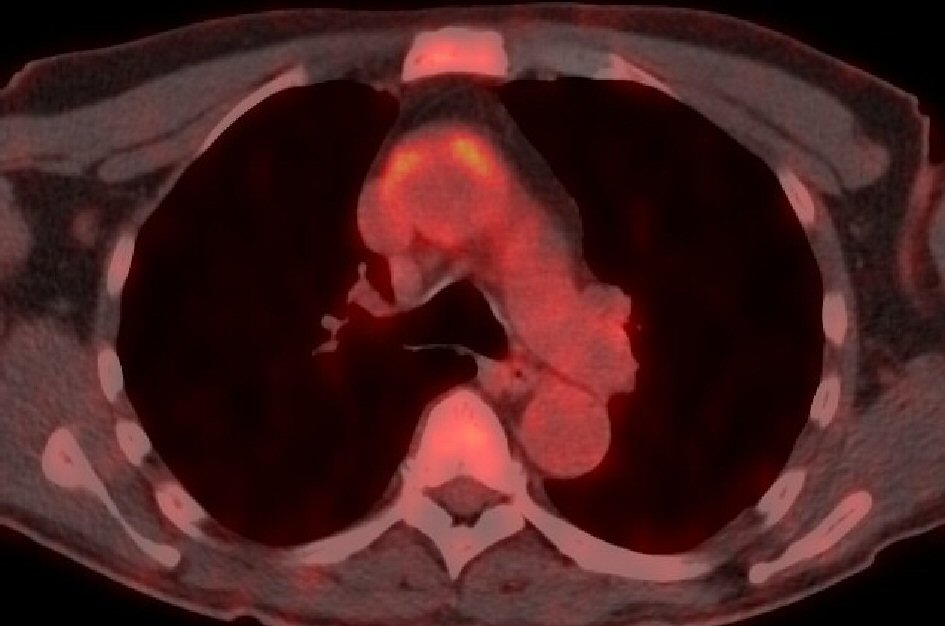 Click for large image | Figure 3. PET scan of patient B reveals an increased up-take of 18F-fludeoxyglucose in the ascending part and arch of the aorta. |
Treatment with 80 mg prednisolone was started which resulted in a dramatic clinical improvement with normalisation of the CRP. Two months after the start of the prednisolone treatment ESR was decreased to 52 mm with a normalisation of the CRP. However, the anemia persisted (Hb 9.2 g/dL with an MCV of 116) with a decrease of the thrombocytes to 116 × 106/mL and 5.8 × 106/mL leucocytes, with only 0.4 × 106/mL monocytes and no circulating blasts. Bone marrow examination revealed normal cellularity with 9% myeloblasts and 25% promonocytes and > 10% dysplastic features in the megakaryopoiesis. There was a normal karyotype. The 9% myeloblasts and 25% promonocytes are consistent with AML. Within a few weeks 16% blasts appeared in the blood. Intensive chemotherapy followed by allogeneic stem cell transplantation resulted in complete remission. A follow-up PET scan revealed no signs of vasculitis.
| Discussion | ▴Top |
We report two patients with vasculitis in relation to malignant myeloid disorders. The first case was a patient with MDS-EB-2 and cutaneous PAN. After immunosuppressive treatment with prednisolone and subsequently azathioprine not only the vasculitis-associated symptoms subsided but there was also a clear improvement of the hematologic and bone marrow findings. In the second patient CMML rapidly developed into AML during successful immunosuppressive treatment with prednisolone for a large vessel vasculitis. Since in this patient no bone marrow examination was performed at diagnosis it is not certain whether the large vessel vasculitis was associated with the CMML or was the presenting symptom of the AML [13]. Whereas these cases provide no pathophysiological evidence, they illustrate the manifestation and course of AIM in MDS and therefore are a good starting point for a discussion on immune system balance in MDS.
In view of the clear association between AIM-like vasculitis and MDS/CMML the question arises whether AIM in MDS is a cause or a consequence. In other words: may the occurrence of an abnormal myeloid progenitor clone lead to lymphocytic dysregulation with subsequent AIM or has the presence or the treatment of AIM per se a pathogenic role in the development of MDS. Or should it be assumed that a dysregulation within the immune system may be responsible for the development of both AIM and MDS? If there is a role of autoimmunity in MDS, it must be argued whether immune dysregulation determines the clinical features (such as the peripheral pancytopenia despite bone marrow hypercellularity) or display a role in the pathogenesis of the disease.
The pathogenesis of MDS is complex and not fully elucidated. A great number of genetic and epigenetic changes may affect the function of the hematopoietic stem cells (HSC) [14, 15]. In addition, there is increasing evidence for a potential contributing role of associated changes in the cells of the microenvironment, e.g. mesenchymal stromal, osteolineage, and endothelial cells, and for alterations in the innate and adaptive immune system [9, 16-18].
The observed immune dysregulation is distinct in low and high risk MDS [9, 17, 19]. In low risk MDS, immune damage of the hematopoietic progenitors is associated with increased number of CD8+ cytotoxic T lymphocytes (CTL), interferon (IFN)-γ producing CD4+, and IL17 producing T helper lymphocytes, as well as increased levels of pleiotropic cytokines such as IFN-γ, TNF-α, IL-6 and TGF-β, with decreased number of regulatory T lymphocytes (Tregs). The epitopes to which the CTLs react are not fully identified. Activated CTL mediate apoptosis of hematopoietic target cells by the perforin/granzyme pathway and by induction of the interaction between Fas-receptor and ligand, where the latter expression is upregulated by IFN-γ, TNF-α and TGF-β. The CTL activity is promoted and maintained by IFN-γ-producing CD4+ and IL17-producing T helper lymphocytes. The immune mediated response induced in this way is facilitated by the decreased number of Tregs, limiting immune evasion by inhibition of hematopoiesis.
In contrast, in high risk MDS, the MDS stem cell may escape tumour surveillance by several immune mediated mechanisms. The key trigger has not yet been identified. Secretion by bone marrow T helper cells of cytokines as IFN-γ and TNF-α may induce the check-point molecules cytotoxic lymphocyte associated protein 4 (CTLA4) and programmed cell death protein 1 (PD-1) in CTLs which may abate the immune mediated cytotoxic capacity. It has been hypothesized that myeloid derived suppressor and mesenchymal stromal as well as dendritic cells directly further abolish the number and activity of natural killer cells and CTLs or by a cytokine (IL-10) mediated increase of the number of Tregs.
The way the above described processes are initiated and whether the effector and target cells are part of the myelodysplastic or normal hematopoietic cells are not fully resolved. Nevertheless, these observations indeed have therapeutic implications. Already more than 20 years ago it was observed that treatment with immunosuppression by means of prednisone not only resulted in remission of the AIM but also may ameliorate the dysplastic features [11]. In low risk MDS immunomodulatory treatment such as anti-thymocyte globulin, ciclosporin, lenalidomide, alemtuzumab, and infliximab may improve the dysplastic features in approximately one-third of the patients [9, 16, 20]. Checkpoint inhibition with CLTA4 inhibitor ipilimumab and PD-1 inhibitors pembrolizumab and nivolumab are widely explored in malignant hematologic diseases, but data in high-risk MDS are limited with so far only modest activity [9, 21, 22]. Treatment with hypomethylating agents such as the nucleoside analogues azacytidine and decitabine leads to a response rate of 20-30% with an additional hematologic improvement in 40% of the MDS patients [23]. These agents do not only inhibit the DNA methyltransferases to activate suppression of certain tumour suppressor genes, but also may display immunological effect by interfering with the action of NK cells, dendritic cells and T cells [24].
In general AIM in patients with MDS/CMML is successfully treated with steroids and other immunomodulatory agents with short-term response rates of up to 80% although many patients relapse or become steroid-dependence [5, 6]. Additional treatment with azacytidine may lead to a high response rate with significant reduction in steroid dose [21]. However, in a review of the treatment efficacy in 55 patients with MDS-associated vasculitis 33% of the reported patients died due either to vasculitis-related or immunosuppressive treatment-related causes [12].
In conclusion, vasculitis may be a presenting symptom of AIM in MDS/CMML. The dysregulation of the innate and adoptive immune system in patients with MDS may be related to the occurrence of AIM. It is not clear whether the treatment of AIM in MDS should be primarily directed against the AIM, to the MDS, or both. Immunosuppressive treatment may not only lead to recovery of the AIM but also to hematological improvement.
Financial Support
No funding or financial support was received.
Author Contributions
YS and LV conceived the idea, JJ and LV wrote the manuscript, EP provided the pathology, and YS, JW and LV edited the manuscript. All authors approved the final version of the manuscript.
Conflict of Interest
All authors disclose no conflict of interest.
Abbreviations
AIM: autoimmune manifestation; AML: acute myeloid leukemia; CMML: chronic myelomonocytic leukemia; CTL: CD8+ cytotoxic T lymphocytes; HSC: hematopoietic stem cell; IFN: interferon; MDS: myelodysplastic syndrome; MDS-EB-2: myelodysplastic syndrome with excess blasts type 2; PAN: polyarteritis nodosa; Treg: regulatory T lymphocytes
| References | ▴Top |
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
doi pubmed - Rhoads CP, Barker WH. Refractory anemia: analysis of one hundred cases. J Am Med Assoc. 1938;110(11):794.
doi - Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51(2):189-199.
doi pubmed - Fozza C. The burden of autoimmunity in myelodysplastic syndromes. Hematol Oncol. 2018;36(1):15-23.
doi pubmed - Wolach O, Stone R. Autoimmunity and inflammation in myelodysplastic syndromes. Acta Haematol. 2016;136(2):108-117.
doi pubmed - Mekinian A, Grignano E, Braun T, Decaux O, Liozon E, Costedoat-Chalumeau N, Kahn JE, et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: a French multicentre retrospective study. Rheumatology (Oxford). 2016;55(2):291-300.
doi pubmed - Grignano E, Mekinian A, Braun T, Liozon E, Hamidou M, Decaux O, Puechal X, et al. Autoimmune and inflammatory diseases associated with chronic myelomonocytic leukemia: A series of 26 cases and literature review. Leuk Res. 2016;47:136-141.
doi pubmed - Giannouli S, Voulgarelis M. A comprehensive review of myelodysplastic syndrome patients with autoimmune diseases. Expert Rev Clin Immunol. 2014;10(12):1679-1688.
doi pubmed - Wang C, Yang Y, Gao S, Chen J, Yu J, Zhang H, Li M, et al. Immune dysregulation in myelodysplastic syndrome: Clinical features, pathogenesis and therapeutic strategies. Crit Rev Oncol Hematol. 2018;122:123-132.
doi pubmed - Komrokji RS, Kulasekararaj A, Al Ali NH, Kordasti S, Bart-Smith E, Craig BM, Padron E, et al. Autoimmune diseases and myelodysplastic syndromes. Am J Hematol. 2016;91(5):E280-283.
doi pubmed - Enright H, Jacob HS, Vercellotti G, Howe R, Belzer M, Miller W. Paraneoplastic autoimmune phenomena in patients with myelodysplastic syndromes: response to immunosuppressive therapy. Br J Haematol. 1995;91(2):403-408.
doi pubmed - Oostvogels R, Petersen EJ, Chauffaille ML, Abrahams AC. Systemic vasculitis in myelodysplastic syndromes. Neth J Med. 2012;70(2):63-68.
pubmed - Chandratilleke D, Anantharajah A, Vicaretti M, Benson W, Berglund LJ. Migratory large vessel vasculitis preceding acute myeloid leukemia: a case report. J Med Case Rep. 2017;11(1):71.
doi pubmed - Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17(1):5-19.
doi pubmed - Ganguly BB, Banerjee D, Agarwal MB. Impact of chromosome alterations, genetic mutations and clonal hematopoiesis of indeterminate potential (CHIP) on the classification and risk stratification of MDS. Blood Cells Mol Dis. 2018;69:90-100.
doi pubmed - Glenthoj A, Orskov AD, Hansen JW, Hadrup SR, O'Connell C, Gronbaek K. Immune mechanisms in myelodysplastic syndrome. Int J Mol Sci. 2016;17(6):944.
doi pubmed - Serio B, Risitano A, Giudice V, Montuori N, Selleri C. Immunological derangement in hypocellular myelodysplastic syndromes. Transl Med UniSa. 2014;8:31-42.
pubmed - Li AJ, Calvi LM. The microenvironment in myelodysplastic syndromes: Niche-mediated disease initiation and progression. Exp Hematol. 2017;55:3-18.
doi pubmed - Ganan-Gomez I, Wei Y, Starczynowski DT, Colla S, Yang H, Cabrero-Calvo M, Bohannan ZS, et al. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia. 2015;29(7):1458-1469.
doi pubmed - Zeidan AM, Stahl M, Komrokji R. Emerging biological therapies for the treatment of myelodysplastic syndromes. Expert Opin Emerg Drugs. 2016;21(3):283-300.
doi pubmed - Fraison JB, Mekinian A, Grignano E, Kahn JE, Arlet JB, Decaux O, Denis G, et al. Efficacy of Azacitidine in autoimmune and inflammatory disorders associated with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Res. 2016;43:13-17.
doi pubmed - Vick E, Mahadevan D. Programming the immune checkpoint to treat hematologic malignancies. Expert Opin Investig Drugs. 2016;25(7):755-770.
doi pubmed - Xie M, Jiang Q, Xie Y. Comparison between decitabine and azacitidine for the treatment of myelodysplastic syndrome: a meta-analysis with 1,392 participants. Clin Lymphoma Myeloma Leuk. 2015;15(1):22-28.
doi pubmed - Lindblad KE, Goswami M, Hourigan CS, Oetjen KA. Immunological effects of hypomethylating agents. Expert Rev Hematol. 2017;10(8):745-752.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.