









| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 7, Number 2, May 2018, pages 72-75
A Metastatic Signet Ring Cell Carcinoma Presented as Acquired Thrombotic Thrombocytopenic Purpura: A Case Report
Noha Eisaa, d, Khalid Nasefb, Ziad Emarahb, Marwa Mohamed Abdel Fattahc, Sameh Shamaab
aHematology Unit, Oncology Center, Mansoura University, Egypt
bMedical Oncology Unit, Oncology Center, Mansoura University, Egypt
cDepartment of Pathology, Faculty of Medicine, Mansoura University, Egypt
dCorresponding Author: Noha Eisa, Hematology Unit, Oncology Center, Faculty of Medicine, Mansoura University, Egypt
Manuscript submitted February 5, 2018, accepted March 1, 2018
Short title: Signet Ring Cell Carcinoma as Acquired TTP
doi: https://doi.org/10.14740/jh386e
| Abstract | ▴Top |
Microangiopathic hemolytic anemia (MAHA) may occur as a paraneoplastic syndrome in some solid tumors, but MAHA accompanied by signet ring cell carcinoma (SRCC) of an unknown origin is very rare. We report a patient who presented with an acute onset of Coombs negative hemolytic anemia and frequent schistocytes in the peripheral blood smear which are typical for MAHA as initial presentation of metastatic SRCC. Our patients fulfilled the criteria of thrombotic thrombocytopenic purpura (TTP) and received the specific treatment for TTP without improvement.
Keywords: Microangiopathic hemolytic anemia; Signet ring cell carcinoma; Thrombotic thrombocytopenic purpura
| Introduction | ▴Top |
Microangiopathic hemolytic anemia (MAHA) refers to any hemolytic anemia related to RBC fragmentation, occurring in association with small vessel disease. In disseminated intravascular coagulopathy (DIC), RBC fragmentation is thought to result from the deposition of fibrin or platelets within the microvasculature. The term “thrombotic microangiopathy (TMA)” is also used to describe syndromes characterized by MAHA, thrombocytopenia, and thrombotic lesions in small blood vessels. The most prominent diagnoses associated with TMA are thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Many different disorders, including preeclampsia, infections, some drug reactions, hematopoietic stem cell transplantation, autoimmune diseases, and malignancies, can cause TMA (i.e., secondary TMA) [1].
Secondary MAHA as a paraneoplastic syndrome in different solid tumors is a serious, often a serious complication of malignancy. Tumor-derived factors, procoagulants, immune complexes, some chemotherapeutic agents, fibrinoid necrosis of bone marrow, and tumor cell emboli of arteries, arterioles, and capillaries are responsible for occurrence of cancer-associated MAHA (CA-MAHA). Chemotherapy is the only effective therapy [2].
MAHA occurs occasionally as a paraneoplastic syndrome in some solid tumors, but MAHA accompanied by signet ring cell carcinoma (SRCC) of an unknown origin is very rare [3]. Herein, we describe a case of SRCC in which MAHA was the initial presentation.
| Case Report | ▴Top |
A 31-year-old male presented with bone aches for more than 2 months followed by easy fatigability in October, 2017 associated with recurrent fever. Upon examination, the patient had severe pallor, jaundice, palpable splenomegaly, but no lymphadenopathy. The patient was admitted to the Hematology Unit, Oncology Center, Mansoura University with laboratory tests showing anemia (Hb 6.6 g/dL; MCV: 92 fL; MCH: 28 pg), normal WBC (5,500/µL), and thrombocytopenia with platelet count 17.6 × 109/L and total bilirubin 3.1 mg/dL (direct 1.4 mg/dL). High lactate dehydrogenase (LDH, 2,137 U/L), with negative viral screen for HCV, HBV and HIV was shown. High serum ferritin (5,052 ng/mL) with negative direct and indirect Coombs test and the pelvi-abdominal ultrasound showed mild hepatosplenomegaly. The blood smear showed frequent schistocytes and polychromatic, nucleated red blood cells and reticulocytosis. Blood coagulation tests were normal to rule out DIC.
According to these clinical and laboratory findings, we planned to start treatment as TTP and continue the essential workup to rule out secondary TTP. We asked for CT of chest, abdomen and pelvis, bone marrow aspiration and biopsy, tumor markers and some immunological markers.
The patient received packed RBCs, pulse steroid for 5 days in parallel with plasma exchange, however, no improvement with deterioration in the performance status and intolerable bone aches which decreased by opioids. Follow-up with laboratory tests showed more rise in LDH, more high serum bilirubin, rising ALT and AST with no improvement of the blood picture and the patient developed two attacks of fits with normal brain CT. Our patient was not responding with more deterioration according to clinical setting and laboratory findings, so we started rituximab 100 mg/m2 once weekly.
The pre- and post-contrast CT revealed a multiple mixed lytic sclerotic lesion seen in all scanned bones, few tiny nodules seen in right lung parenchyma measuring 6 mm mostly metastatic, mild enlarged liver with multiple non-enhancing focal lesions, largest 4 × 7 mm.
Among the tumor markers that were assessed, both carcinoembryonic antigen level (807.0 ng/mL) and cancer antigen 19-9 (80 U/mL) were elevated. The subsequent bone marrow biopsy revealed bone marrow infiltrated by malignant tumor tissue in the form of malignant acini lined by malignant epithelial cells that show moderate atypia and pleomorphism, and scattered signet ring cells are also seen in Figures 1 and 2. Immunostaining revealed positive staining for CK in neoplastic cells.
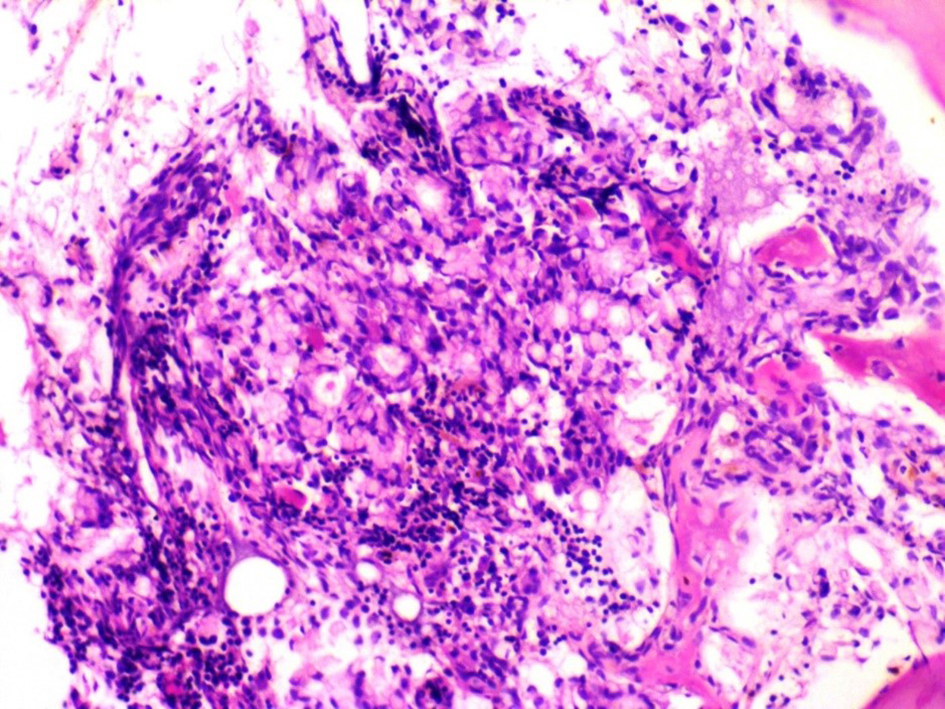 Click for large image | Figure 1. Metastatic carcinoma in the form of signet ring cells seen admixed within a background of bone marrow hemopoietic elements. |
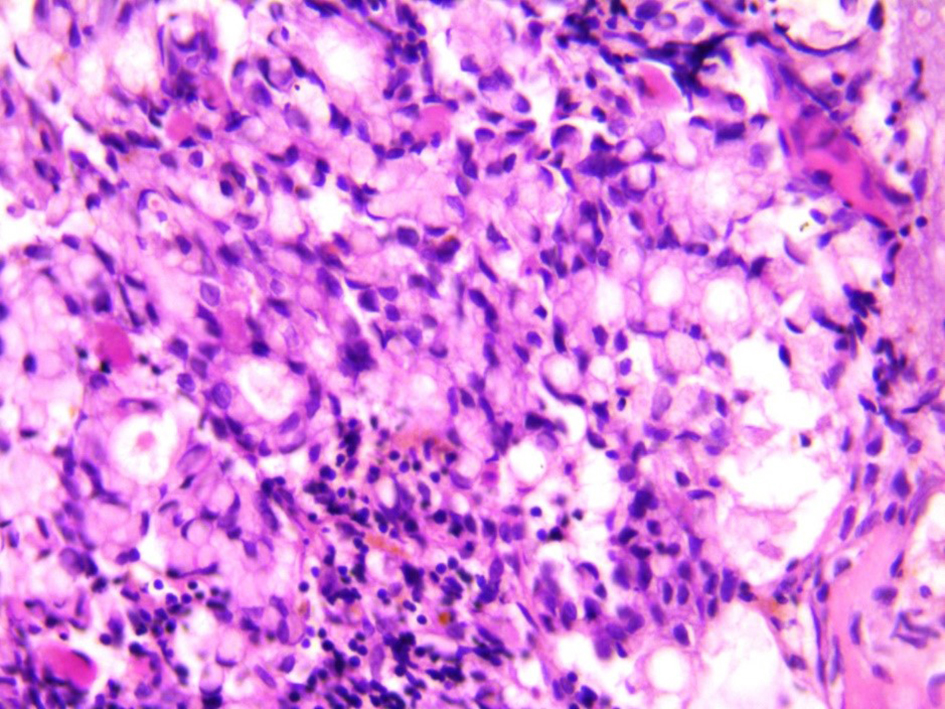 Click for large image | Figure 2. Scattered aggregates of signet ring carcinoma cells seen within the bone marrow elements (× 40). |
After these results, the patient referred to Medical Oncology ward with poor general condition, feverish with high liver function tests (total bilirubin: 13.3 mg/dL, ALT: 610.37 UI/L, AST: 349.56 UI/L), anemia (Hb 6.5 g/dL), and thrombocytopenia with platelet count 12 × 109/L.
The patient planned for CT-guided biopsy and upper GIT endoscopy after improvement to detect the primary site but unfortunately, the patient continued to deteriorate despite repeated blood transfusions, antibiotics and had been expired 2 days later.
| Discussion | ▴Top |
Primary TMA syndromes are specific disorders that require specific treatments. They include TTP, HUS, and drug-induced TMA. Although these disorders have been studied separately in the past, in recent decades, they have been combined as common TMA syndrome, which includes definitive clinical and pathological features, MAHA, thrombocytopenia, and organ injury [4]. CR-MAHA is distinguished from primary TMA in terms of its underlying disorders and treatment options. CR-MAHA has been reported in association with several types of carcinoma since the 1980s. Lechner et al conducted a literature review of 168 reports of CR-MAHA; according to cancer type, gastric cancer was the most frequent, followed by breast, prostate, and lung cancers [5].
A Korean study reported that 14 (25.5%) out of 55 MAHA patients had gastric cancer [6]. In our patient, MAHA was the initial finding of SRCC in the bone marrow; however, we did not reach the primary site due to rapid deterioration in general condition. CR-MAHA is a rare and fatal complication of malignant tumors. Among patients with bone marrow metastases from cancer, MAHA patients have a worse prognosis than patients without MAHA [7]. Our patients had expired 22 days after initial presentation which is parallel with the different literature that proved most of CR-MAHA cases die within few weeks, majority with infection as final insult [8, 9].
Our patient was diagnosed with CA-MAHA on the basis of the findings of bone marrow examination and CT conducted 7 days after the diagnosis of MAHA. Many studies have recommended a thorough tumor workup, including bone marrow examination, in cases of progressive MAHA of unknown origin [6].
There are some clinical symptoms and signs which are more prominent in CA-MAHA than in non-CA-MAHA like bone pains and respiratory symptoms [8, 10]. Actually our patient experienced both significant bone pain and dyspnea.
Although several studies have suggested that fibrinoid necrosis of the bone marrow and tumor cell emboli of the arteries, arterioles, and capillaries are the causes of CA-MAHA, the pathogenesis of CA-MAHA remains unclear [11, 12]. Some studies have reported a decrease in the level of the von Willebrand factor (vWF)-cleaving protease, and ADAMTS13 plays a role in CA-MAHA [8]. We were unable to evaluate the presence of tumor emboli of the arteries, ADAMTS13, or tumor necrosis factor alpha (TNF-α) in this case because the patient expired rapidly.
SRCC is a subtype of mucin-producing adenocarcinomas. However, mucin-producing adenocarcinomas and SRCC show clinicopathological differences [13, 14]. Our patient is a 31-year-old male with the fact that the gastrointestinal tract SRCC is more common in the young and in males and has a worse prognosis than mucin-producing adenocarcinomas. SRCC is frequently complicated by metastases to the regional lymph nodes, peritoneal surfaces, ovaries, lungs, and bone marrow [15].
There is no definitive treatment of choice for CA-MAHA. The low platelet count and hemoglobin level make red blood cell and platelet transfusion obligatory. Actually in our patient, we started urgent treatment for TTP in the form of plasma exchange and pulse steroids with no response, then second line added in the form of rituximab; however, he showed progression because the treatment of CA-MAHA differed than that of non-CA-MAHA. And the optimal treatment is to give the specific chemotherapy protocols with supportive blood transfusion. An additional heparinization and administration of glucocorticoids are subjects of controversial discussion. The main focus of treatment is to reduce the tumor mass. A chemotherapy should be started as soon as possible after diagnosis [6, 16-19].
Conclusions
CR-MAHA is an uncommon paraneoplastic syndrome with rapidly serious outcome. Its association with carcinoma specifically signet ring cell variant deliberates a worse prognosis. Consequently, laboratory findings which suggest MAHA make it necessary to search for the primary carcinoma as early as possible as aggressive tumor control is possibly the most effective treatment for CR-MAHA as well.
Conflict of Interest
The authors have no conflict of interest.
Disclaimers
The submitted article is our own and not an official position of the institution or funder.
| References | ▴Top |
- Morishita E. [Diagnosis and treatment of microangiopathic hemolytic anemia]. Rinsho Ketsueki. 2015;56(7):795-806.
pubmed - Yuce T, Bakkaloglu O, Kose M, Akpinar T, Tukek T. Microangiopathic hemolytic anemia in metastasized signet ring cell carcinoma: a report of three cases. International Journal of Hematology Research. 2016;2(2):136-138.
doi - Shin SY, Park H, Chae SW, Woo HY. Microangiopathic hemolytic anemia as the first manifestation of metastatic signet ring cell carcinoma of unknown origin: a case report and review of literature. Korean J Lab Med. 2011;31(3):157-161.
doi pubmed - George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666.
doi pubmed - Lechner K, Obermeier HL. Cancer-related microangiopathic hemolytic anemia: clinical and laboratory features in 168 reported cases. Medicine (Baltimore). 2012;91(4):195-205.
doi pubmed - Abdel Samie A, Sandritter B, Theilmann L. [Severe microangiopathic hemolytic anemia as first manifestation of a CUP syndrome. Rapid hematologic remission under polychemotherapy]. Med Klin (Munich). 2004;99(3):148-153.
doi pubmed - Etoh T, Baba H, Taketomi A, Nakashima H, Kohnoe S, Seo Y, Fukuda T, et al. Diffuse bone metastasis with hematologic disorders from gastric cancer: clinicopathological features and prognosis. Oncol Rep. 1999;6(3):601-605.
doi - Francis KK, Kalyanam N, Terrell DR, Vesely SK, George JN. Disseminated malignancy misdiagnosed as thrombotic thrombocytopenic purpura: A report of 10 patients and a systematic review of published cases. Oncologist. 2007;12(1):11-19.
doi pubmed - Hahn JS, Lee DH, Lee SJ, Min YH, Ko YW. A clinical study on microangiopathic hemolytic anemia. Korean J Hematol. 1991;26:263-279.
- Oberic L, Buffet M, Schwarzinger M, Veyradier A, Clabault K, Malot S, Schleinitz N, et al. Cancer awareness in atypical thrombotic microangiopathies. Oncologist. 2009;14(8):769-779.
doi pubmed - Hilgard P, Gordon-Smith EC. Microangiopathic haemolytic anaemia and experimental tumour-cell emboli. Br J Haematol. 1974;26(4):651-659.
doi pubmed - Murgo AJ. Thrombotic microangiopathy in the cancer patient including those induced by chemotherapeutic agents. Semin Hematol. 1987;24(3):161-177.
pubmed - Kim KS, Kim YD, Han KH, Lee SH, Kim JH, Choi HY, Cheon GJ, et al. Endoscopic findings and clinicopathological characteristics of signet ring cell carcinoma of the stomach. Korean J Med. 2007;73:596-602.
- Akamatsu S, Takahashi A, Ito M, Ogura K. Primary signet-ring cell carcinoma of the urinary bladder. Urology. 2010;75(3):615-618.
doi pubmed - Tot T. Cytokeratins 20 and 7 as biomarkers: usefulness in discriminating primary from metastatic adenocarcinoma. Eur J Cancer. 2002;38(6):758-763.
doi - Lee JL, Lee JH, Kim MK, Cho HS, Bae YK, Cho KH, Bae SH, et al. A case of bone marrow necrosis with thrombotic thrombocytopenic purpura as a manifestation of occult colon cancer. Jpn J Clin Oncol. 2004;34(8):476-480.
doi pubmed - Jeong GY, Yoon HR, Kim SH. A case of bone marrow necrosis preceeding acute monoblastic leukemia. Korean J Clin Pathol. 1999;19:172-176.
- Arkenau HT, Mussig O, Buhr T, Jend HH, Porschen R. Microangiopathic hemolytic anemia (MAHA) as paraneoplastic syndrome in metastasized signet ring cell carcinomas: case reports and review of the literature. Z Gastroenterol. 2005;43(8):719-722.
doi pubmed - Ortega Marcos O, Escuin F, Miguel JL, Gomez Fernandez P, Perez Fontan M, Selgas R, Sanchez Sicilia L. Hemolytic uremic syndrome in a patient with gastric adenocarcinoma: partial recovery of renal function after gastrectomy. Clin Nephrol. 1985;24(5):265-268.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.