

| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Original Article
Volume 7, Number 2, May 2018, pages 43-50
Retrospective Study of High Hemoglobin Levels in 56 Young Adults
Alexandra Desnoyersa, Michel Pavica, Paul-Michel Houlea, b, Jean-Francois Castillouxa, Patrice Beauregarda, Line Delislea, Richard Le Blanca, Jean Dufresnea, Josie-Anne Boisjolya, Vincent Ethiera
aSherbrooke University Hospital Center, Sherbrooke, QC, Canada
bCorresponding Author: Paul-Michel Houle, University of Sherbrooke, Internal Medicine Common Core, 751 rue des Capucines, Apartment 304, Sherbrooke, QC J1E 0G5, Canada
Manuscript submitted January 18, 2018, accepted February 19, 2018
Short title: High Hemoglobin Levels in 56 Young Adults
doi: https://doi.org/10.14740/jh375w
| Abstract | ▴Top |
Background: Erythrocytosis is a frequent request for consultation in the hematologic field. The diagnostic approach is well established in the general population but in a young adult, finding the etiology of erythrocytosis can be a real diagnostic challenge.
Methods: This is an observational retrospective unicentric study made at the Sherbrooke University Hospital Center, over a period of 20 years (1995 - 2015). Every patient aged between 16 and 35 years old with a significant elevation of hemoglobin or hematocrit was included (hemoglobin > 185 g/L and/or hematocrit > 0.52 in men; hemoglobin > 165 g/L and/or hematocrit > 0.48 in women).
Results: Totally, 426 patients met the inclusion criteria (over a total of 113,453 complete blood counts) but only 56 entered the study for investigations. The majority of patients were of male gender, 43% of the patients were obese, 59% were smokers and 38% used excess alcohol or recreational drugs. Twenty-five patients had the diagnosis of absolute erythrocytosis. Seven patients had the diagnosis of relative erythrocytosis and no cause could be identified in 24 patients. No primary erythrocytosis was found in this cohort. Among the 25 patients with secondary erythrocytosis, hypoxia was the most frequent etiology identified. Less than half of the patients in the cohort had long term follow-up. Search for JAK2 mutation and serum EPO dosage were performed in 17.9% and 23.2% of cases respectively. Seven patients were treated with aspirin and five patients had phlebotomies.
Conclusions: This retrospective study reveals an actual clinical management that is often discordant with the current recommendations and a frequent lack of follow-up after initial investigations. Harmonization of management of erythrocytosis appears to be highly desirable.
Keywords: Hematology; Erythrocytosis; Young adults; Epidemiology
| Introduction | ▴Top |
Erythrocytosis represents a frequent request for consultation in the hematologic field. Frequently, it is an incidental finding secondary to a persistent elevation of hemoglobin and hematocrit. Erythrocytosis is suspected when repeated values of hemoglobin are above 165 g/L in female and 185 g/L in male, or when hematocrit values exceed 48% in female and 52% in male. However, the term absolute erythrocytosis is used when an erythrocytic mass exceeds 125% of the predicted value [1-4]. When the erythrocytic mass is not increased, erythrocytosis is said to be “relative” [5].
Erythrocytosis is subsequently classified according to its etiology, whether primary or secondary. Primary erythrocytosis is due to a direct defect within the red blood cell (RBC) progenitors [3]. Secondary erythrocytosis is due to external factors stimulating the RBC production. The latter type is more common in the general population [6]. Its differential diagnosis is broad and encompasses notably cardio-pulmonary pathologies, smoking and exogenous androgens. (Table 1, [1, 3, 4, 7-18]) It is important to use a systematic approach to identify the underlying etiology and to manage it accordingly. Many diagnostic algorithms have been proposed in the literature [19]. Proper management depends on the underlying cause. Phlebotomies may be proposed to patients presenting with hyperviscosity related symptoms [20] (Fig. 1).
 Click to view | Table 1. Main Etiologies of Erythrocytosis ([1, 3, 4, 7-18]) |
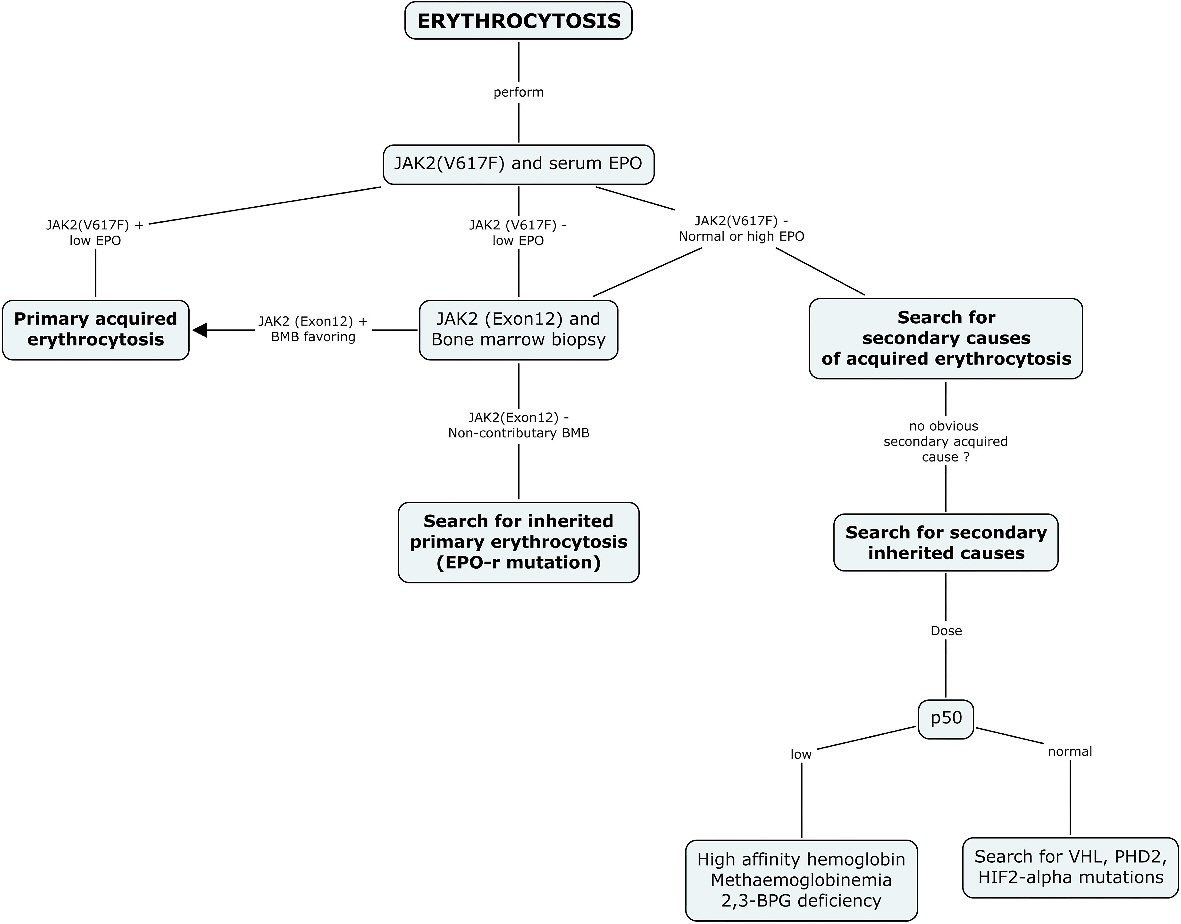 Click for large image | Figure 1. Diagnostic algorithm suggested for erythrocytosis. The current diagnostic approach recommended for the investigation of erythrocytosis systematically includes a search for JAK2 (V617F) mutation and serum EPO as the first step in every patient. A positive JAK2 (V617F) result should orient the diagnosis and management towards a diagnosis of primary acquired erythrocytosis. A normal or high EPO should guide the clinician towards the search for a secondary cause of acquired erythrocytosis, and when no cause is found, the search for a secondary inherited cause. A negative result for JAK2 (V617F) mutation and a low EPO level should be further investigated with a bone marrow biopsy and a search for JAK2 (exon12), and when non-contributory, should prompt a search for inherited primary erythrocytosis. EPO: erythropoietin; BPG: bisphosphoglycerate; HIF-2: hypoxia inducible factor 2; PHD2: prolyl hydroxylase domain protein 2; VHL: von Hippel-Lindau. |
Even though general etiological profile and clear management indications are already established in medical literature, data is still lacking regarding the etiologic profile on the population level, especially in young adults. The purpose of this study was to answer this very question by interrogating the population of young adults at the Sherbrooke University Hospital Center (CHUS), in Quebec. The secondary outcome was to compare the actual clinical management of these patients with the current recommendations in the literature.
| Methods | ▴Top |
Study design
In this observational study design, every case of erythrocytosis identified at the CHUS between January 1, 1995 and December 31, 2015, was examined retrospectively through computerized medical files. Patients had to be between 16 and 35 years old at the time of the erythrocytosis diagnosis. Within this specified window, patients had to have at least one complete blood count performed at the CHUS with a hemoglobin value of 185 g/L or higher for men and 165g/L or higher for women or a hematocrit value of 52% or higher for men and of 48% or higher for women. The protocol of this study was approved by the institutional scientific and ethical committees.
Patients
All patients were aged between 16 and 35 years old and had at least one criteria of suspicion for erythrocytosis at one time during the specified window. After the review of every collected file, patients were excluded if they didn’t have their investigation and/or follow-up for the erythrocytosis performed at the CHUS and if the computerized medical files contained no more information than the initial complete blood count. Based on the available data, a consensus was reached between the main authors for cases whose inclusion in the cohort was debatable.
Data collection
Data collected in the computerized medical files were: anthropometric measurements, comorbidities, laboratory tests (dates and results) and imaging tests (dates and results). Additional data were collected through manual extraction of the medical files and consisted of the following: lifestyle habits (tobacco use, alcohol intake and recreational drug use), medications (androgens, steroids, and erythropoietin), data on medical follow-up and treatments offered.
Variable and objectives
The main outcome of this study was to establish the etiological profile of erythrocytosis among the population of young adults at a Canadian university hospital center. The etiology was determined following an analysis of the medical files. When no etiology was clearly established during follow-up, patients were classified according to the most likely etiology determined by a consensus among the main authors. Sometimes, no cause could be identified even after thorough review of the medical files. The secondary outcome was to evaluate the management of these patients and compare it to the current recommendations in the medical literature, with the purpose of improving the quality of care.
Statistical analyses
Statistical analyses were of a descriptive type in this study. Categorical variables were described using frequencies and proportions. Continuous variables were described using means and standard deviation if they were normal in type or using medians and interquartile ranges if normality was not respected.
| Results | ▴Top |
Patients
A total of 113,453 complete blood counts were performed in the young adult population at the CHUS between January 1995 and December 2015. Among these, 426 patients met the criteria for suspicion of erythrocytosis. Within that number, 56 patients were selected for the study, having been investigated and monitored for their erythrocytosis.
The majority of these patients were male. In this cohort, 43% were obese and a third of these had a morbid obesity (class III obesity). As for lifestyle habits, 59% of patients were smokers and 38% were taking alcohol (11%) or using recreational drugs (27%) daily (Tables 2-4).
Click to view | Table 2. Distribution of Cases per Year |
Click to view | Table 3. Demographic and Clinical Characteristics of Patients |
Click to view | Table 4. Erythrocytosis Criteria |
Etiological data
Even with a preselection of patients being investigated for erythrocytosis detected with a complete blood count, no cause could be identified for 24 of these young adults. Even after a complete review of the computerized medical files by the authors, the diagnosis still remained uncertain. A diagnosis of relative erythrocytosis was made in seven cases. As for the remaining 25 cases of absolute erythrocytosis in the cohort, no case of primary erythrocytosis was found. Thus, there was no case of myeloproliferative syndrome, including polycythemia vera (PV), for the period studied in the young adults at the CHUS (Table 5).
Click to view | Table 5. Major Etiologies of Erythrocytosis |
The 25 cases of absolute erythrocytosis had a secondary etiology. Of these 25 patients, the diagnosis was either clearly identified in the medical records or attributed as most likely after a review of the records by the authors. The two hereditary cases were diagnosed as high oxygen affinity hemoglobin. No genetic mutation was found among patients for whom genetic mutation tests were done, which represents 17.9% of the cohort. Hypoxia was the most frequent extrinsic factor responsible for erythrocytosis [21] in our population of young adults. Of these 13 patients, four had a heart disease, five had a lung disease and eight were smokers. No tumour secreting erythropoietin was found within this cohort. Exogenous androgens prescribed in the context of transgender hormonal therapy were responsible for the four cases of absolute erythrocytosis (Table 6).
Click to view | Table 6. Detailed Etiologies of Secondary Erythrocytosis Cases |
Secondary outcomes
Every patient included in this study was questioned and a physical exam was performed following the discovery of erythrocytosis. Almost half of these patients (n = 26) were not evaluated by a specialist following the discovery of an abnormal hemoglobin or hematocrit value in the complete blood count. Five patients were dismissed after a single visit with a specialist (four with hematologists and one with pneumologist). For the 25 patients who had long-term follow-up, the median length was 42 months. Among these 25 patients, 10 had a follow-up with a hematologist and six with an internist. The other patients had a follow-up in the medical speciality related to their other comorbidities; either in cardiology, pneumology, nephrology or endocrinology.
The JAK2 mutation assay and the serum erythropoietin dosage were performed in 17.9% and 23.2%, respectively. The JAK2 mutation assay, which is available at the CHUS since 2005, was performed in 10 of the 31 patients investigated after 2005 (32.3%). In the majority of patients, an investigation for secondary causes seems to have been performed with complementary laboratory and imaging tests, such as urine analysis, liver function tests, chest radiography and abdominal ultrasound. Although two cases of high oxygen affinity hemoglobin were diagnosed in the context of a positive family history, measurement of P50 was never performed (Table 7).
Click to view | Table 7. Patients Management |
Aspirin was prescribed to seven patients following erythrocytosis diagnosis. Five patients had phlebotomies for their symptoms. No death due to erythrocytosis occurred during the follow-up period. Only one patient had a major complication attributable to blood hyperviscosity. This patient was monitored in cardiology for cyanotic heart disease with known as secondary erythrocytosis, but untreated. This patient developed a pulmonary artery thrombosis and was subsequently treated with phlebotomies and a long-term anticoagulation with Coumadin. Hemoglobin and hematocrit values at the time of the thrombotic event were 202 g/L and 0.624, respectively. One patient had hemoptysis in the context of pneumonia and his hemoglobin and hematocrit values at the time were in the normal range at 175 g/L and 0.515 respectively. No association between hemoptysis and erythrocytosis was made by the specialists who evaluated this patient. Four cases of gout arthritis occurred, without any clear association with the underlying erythrocytosis.
| Discussion | ▴Top |
The main conclusion emerging from this study is that erythrocytosis in young adults is predominantly secondary to an external factor that stimulates erythropoiesis. Poor lifestyle habits such as smoking and obesity are over-represented in this particular population in comparison with the general population. The percentage of obese patients (43%) is more than twice the one of the general population which was 18% (self-reported) in the province of Quebec in 2014 [22]. In the study, 59% patients were smokers, which is far more than what is reported in the general population which was 19.6% in the province of Quebec in 2014 [23]. More than half of the reported cases of erythrocytosis were actually related to hypoxia, which suggests that poor lifestyle habits have a significant etiological role.
Conversely, no case of primary erythrocytosis was diagnosed in this population. The prevalence of PV in the general population reported in the medical literature is 44 to 57 per 100,000 people [7]. The incidence of PV in childhood and adolescence is extremely rare. Only about 50 cases of pediatric PV have been reported in all the medical literature [24]. An epidemiological study made in United Kingdom found an incidence of 0.49 cases of PV per 100,000 people among adults aged between 15 and 34 [25]. This data may explain, at least partially, the absence of PV in our cohort.
Furthermore, it should be noted that no cause could be identified in a large proportion of patients despite follow-up and management by a specialist. The prevalence of idiopathic erythrocytosis in the general population is estimated to be 110 per 100,000 people [26, 27]. The prevalence in our cohort was lower, with 21 cases per 100,000 people. Among these cases of idiopathic erythrocytosis, nearly a third of patients were hospitalised in a psychiatric unit at the time of the first complete blood count. A decompensation of the underlying psychiatric illness and deliberate intoxications with drugs or medications were frequently mentioned as the reasons for admission on the hospitalisation summary. This suggests that some of these patients probably had a relative erythrocytosis.
The etiological profile of erythrocytosis in young adults that has been revealed in this study sheds a new light over the distribution of the different causes in this particular population. In the future, this would allow us to better guide the diagnosis and management of this entity. In our opinion, hereditary erythrocytosis deserves mention in the differential diagnosis, although it should be kept in mind that it represents only a small minority of the etiologies in this population, unlike the acquired causes of erythrocytosis.
This study also highlighted the fact that a rise in hemoglobin is often trivialized. Indeed, more than half of these young adults had no referral or follow-up by any specialist. It should also be noted that of the 426 records of young adults who met the inclusion criteria that the computer program identified, 370 had to be rejected because of an incomplete record or lack of management of erythrocytosis. Even when a follow-up was undertaken, the management was often disorganised and discordant with the algorithms suggested in the literature. A secondary cause for erythrocytosis was often assumed but weakly proven. In our opinion, the search for JAK2 mutation as well as serum erythropoietin dosage should be performed in the very beginning of investigations. Less than one in four patients had those tests performed in this study. Even after it became available in 2005 at the CHUS, the JAK2 mutation was searched only in a third of patients with erythrocytosis. This fact could partially explain the absence of primary absolute erythrocytosis diagnosed in our study. No other genetic mutations that could be associated with elevated hemoglobin were investigated. Even the P50 was never measured. The two cases of high oxygen affinity hemoglobin were diagnosed based on family history, but not confirmed. Laboratory and imaging tests were requested in patients as part of their long-term follow-up, probably in search of a secondary cause of erythrocytosis or in the assessment of their underlying disease. This study therefore reveals the lack of structure in the diagnostic approach and follow-up of patients presenting with erythrocytosis when compared with the current recommendations. However, the diagnostic approach and criteria have evolved during the last 20 years. From 1970 to 2001, the criteria used for the diagnosis of PV were those of the Study Group on Polycythemia. These criteria were revised for the first time in 2001 by the World Health Organisation (WHO) [28]. One of the hypotheses that can explain the disparate care between the patients is the evolution of diagnostic criteria and the availability of new tests during the period under study.
In light of the results of our study, we suggest a new management algorithm adapted to the etiological profile of the young adult (Fig. 2).
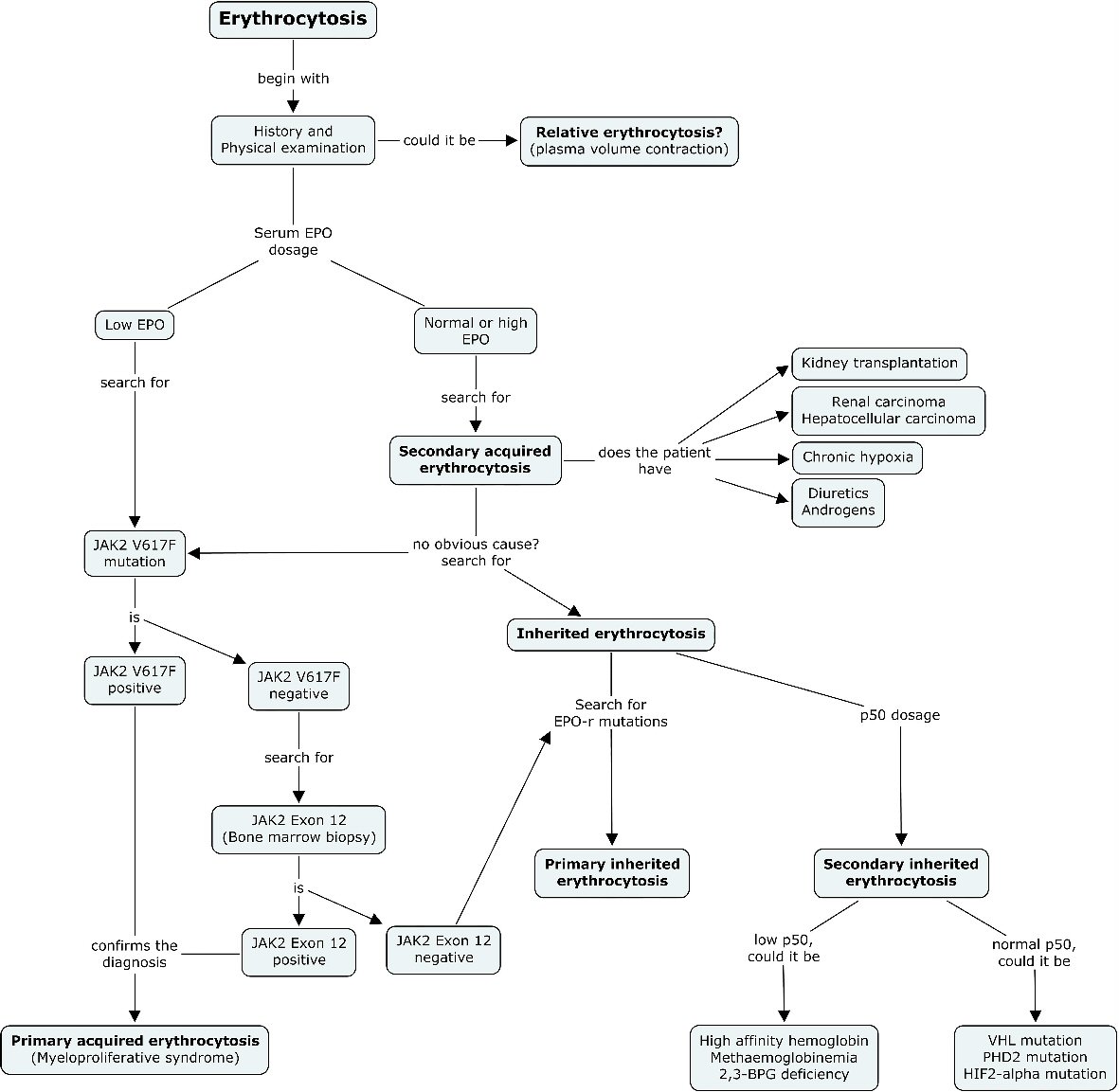 Click for large image | Figure 2. New diagnostic algorithm suggested for erythrocytosis. After exclusion of relative erythrocytosis, serum erythropoietin dosage should be undertaken early in the diagnostic course before concluding of a secondary cause for erythrocytosis in the young adult. In the case of low EPO, JAK2 (V617F) mutation should systematically be searched. A bone marrow biopsy for JAK2 exon 12 search should be undertaken with a negative JAK2 (V617F) mutation. In the case of normal or high EPO, and when no obvious cause for secondary acquired erythrocytosis could be found, a search for inherited erythrocytosis should be undertaken. A search for EPO-r mutations and P50 dosage must then be taken into consideration to complete the diagnostic evaluation. |
Conclusions
To the best of our knowledge, this is the first study to investigate the etiological profile of erythrocytosis in the young adults (16 - 35 years old). Despite its retrospective nature, it reveals a management that is often discordant with the current algorithms recommended in the literature for management of erythrocytosis at all ages and a frequent absence of follow-up after the initial investigations. Hereditary erythrocytosis deserves to be systematically mentioned in this age group, although it represented only a minority of cases in our cohort. Secondary acquired causes of erythrocytosis were the etiologies most frequently diagnosed, whereas myeloproliferative syndromes were not represented in our study. The symptoms which can become important and the thrombotic and hemorrhagic complications associated with blood hyperviscosity require a regular follow-up, especially in a young population [29]. We suggest that modifications be made in the diagnostic approach and management of erythrocytosis in this population. We adapted the current algorithm in light of the results of this study and believe it might better guide clinical decisions in the future.
Acknowledgments
This work was supported by the Research Center of the Sherbrooke University Hospital Center.
Author Contribusions
AD and MP designed the search protocol. AD collected and analysed the data. AD and MP wrote the manuscript. PMH translated the manuscript. JFC, PB, LD, RL, JD, JAB and VE contributed to data collection and analysis.
Conflict of Interest
The authors do not declare any conflict of interest.
| References | ▴Top |
- Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-951.
doi pubmed - Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
doi pubmed - Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14-22.
doi pubmed - Busque L, Porwit A, Day R, Olney HJ, Leber B, Ethier V, Sirhan S, et al. Laboratory investigation of Myeloproliferative Neoplasms (MPNs): recommendations of the Canadian Mpn group. Am J Clin Pathol. 2016;146(4):408-422.
doi pubmed - Lawrence JH, Berlin NI. Relative polycythemia; the polycythemia of stress. Yale J Biol Med. 1952;24(6):498-505.
pubmed - Lee G, Arcasoy MO. The clinical and laboratory evaluation of the patient with erythrocytosis. Eur J Intern Med. 2015;26(5):297-302.
doi pubmed - Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55(3):595-600.
doi pubmed - Stein BL, Moliterno AR, Tiu RV. Polycythemia vera disease burden: contributing factors, impact on quality of life, and emerging treatment options. Ann Hematol. 2014;93(12):1965-1976.
doi pubmed - Titmarsh GJ, Duncombe AS, McMullin MF, O’Rorke M, Mesa R, De Vocht F, Horan S, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol. 2014;89(6):581-587.
doi pubmed - Al-Sheikh M, Mazurier E, Gardie B, Casadevall N, Galacteros F, Goossens M, Wajcman H, et al. A study of 36 unrelated cases with pure erythrocytosis revealed three new mutations in the erythropoietin receptor gene. Haematologica. 2008;93(7):1072-1075.
doi pubmed - Arcasoy MO, Karayal AF. Erythropoietin hypersensitivity in primary familial and congenital polycythemia: role of tyrosines Y285 and Y344 in erythropoietin receptor cytoplasmic domain. Biochim Biophys Acta. 2005;1740(1):17-28.
doi pubmed - Esparcieux A, Francina A, Vital-Durand D. [Abnormal hemoglobins with high oxygen affinity in the differential diagnosis of polycythemia]. Rev Med Interne. 2011;32(10):e105-107.
doi pubmed - Fermo E, Bianchi P, Vercellati C, Marcello AP, Garatti M, Marangoni O, Barcellini W, et al. Recessive hereditary methemoglobinemia: two novel mutations in the NADH-cytochrome b5 reductase gene. Blood Cells Mol Dis. 2008;41(1):50-55.
doi pubmed - Yilmaz D, Cogulu O, Ozkinay F, Kavakli K, Roos D. A novel mutation in the DIA1 gene in a patient with methemoglobinemia type II. Am J Med Genet A. 2005;133A(1):101-102.
doi pubmed - Galacteros F, Rosa R, Prehu MO, Najean Y, Calvin MC. [Diphosphoglyceromutase deficiency: new cases associated with erythrocytosis]. Nouv Rev Fr Hematol. 1984;26(2):69-74.
pubmed - Percy MJ, Beard ME, Carter C, Thein SL. Erythrocytosis and the Chuvash von Hippel-Lindau mutation. Br J Haematol. 2003;123(2):371-372.
doi pubmed - Wilson R, Syed N, Shah P. Erythrocytosis due to PHD2 mutations: a review of clinical presentation, diagnosis, and genetics. Case Rep Hematol. 2016;2016:6373706.
- Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, Lee FS. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358(2):162-168.
doi pubmed - Barba T, Boileau JC, Pasquet F, Hot A, Pavic M. [Inherited primitive and secondary polycythemia]. Rev Med Interne. 2016;37(7):460-465.
doi pubmed - Keohane C, McMullin MF, Harrison C. The diagnosis and management of erythrocytosis. BMJ. 2013;347:f6667.
doi pubmed - Furukawa T, Narita M, Sakaue M, Otsuka T, Kuroha T, Masuko M, Azegami T, et al. Primary familial polycythaemia associated with a novel point mutation in the erythropoietin receptor. Br J Haematol. 1997;99(1):222-227.
doi pubmed - Statistics Canada. Overweight and obese adults (self-reported), 2014. 82-625-X. 2016.
- Statistics Canada. Smokers by sex, provinces and territories (Percent). 2016.
- Cario H, McMullin MF, Bento C, Pospisilova D, Percy MJ, Hussein K, Schwarz J, et al. Erythrocytosis in children and adolescents-classification, characterization, and consensus recommendations for the diagnostic approach. Pediatr Blood Cancer. 2013;60(11):1734-1738.
doi pubmed - McNally RJ, Rowland D, Roman E, Cartwright RA. Age and sex distributions of hematological malignancies in the U.K. Hematol Oncol. 1997;15(4):173-189.
doi - Randi ML, Bertozzi I, Cosi E, Santarossa C, Peroni E, Fabris F. Idiopathic erythrocytosis: a study of a large cohort with a long follow-up. Ann Hematol. 2016;95(2):233-237.
doi pubmed - Ruggeri M, Tosetto A, Frezzato M, Rodeghiero F. The rate of progression to polycythemia vera or essential thrombocythemia in patients with erythrocytosis or thrombocytosis. Ann Intern Med. 2003;139(6):470-475.
doi pubmed - Briere J. Revision des criteres diagnostiques de l’OMS pour les PV, TE et MPF. Hematologie. 2008;14(3):208-15.
- Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F, Randi ML, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874-1881.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.