


| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 3, Number 2, June 2014, pages 57-60
Epstein-Barr Virus Associated Hemophagocytic Lymphohistiocytosis Presenting as Fever of Unknown Origin With Fatal Outcome
Mansoor C. Abdullaa, c, Jemshad Alungala, Lekha R. Nairb, Sujathan Pa
aDepartment of General Medicine, M.E.S. Medical College, Perinthalmanna, Kerala 679338, India
bDepartment of Pathology, M.E.S. Medical College, Perinthalmanna, Kerala 679338, India
cCorresponding Author: Mansoor C. Abdulla, Department of General Medicine, M.E.S. Medical College, Perinthalmanna, Kerala 679338, India
Manuscript accepted for publication April 1, 2014
Short title: Hemophagcytic Lymphohistiocytosis
doi: https://doi.org/10.14740/jh134w
| Abstract | ▴Top |
Epstein-Barr virus (EBV) associated hemophagocytic lymphohistiocytosis (HLH) is a rare and aggressive disease having a high mortality rate without early and effective therapy. In this report, we are sharing our experience of a 24-year-old unmarried lady with fever of unknown origin (FUO) who had a fatal form of HLH due to EBV infection. Since HLH is a potentially fatal hyperinflammatory condition, its prompt identification is prudent for early and effectual treatment.
Keywords: Epstein-Barr virus; Hemophagocytic lymphohistiocytosis; Fever of unknown origin
| Introduction | ▴Top |
Epstein-Barr virus (EBV) associated hemophagocytic lymphohistiocytosis (HLH) is an infrequent complication characterized by altered immune response and hypercytokinemia, characterized clinically by fever, splenomegaly and cytopenia accompanied by histological evidence of hemophagocytosis, resulting in extremely high serum levels of ferritin, lactate dehydrogenase and soluble CD25. Clinical presentations of EBV are diverse ranging from asymptomatic carrier state to a fatal overwhelming infection. HLH can be associated with various infections, autoimmune disorders or malignancies. Viruses of herpes family especially EBV are frequently encountered as etiological agents. Early identification and appropriate therapies are most essential as it causes a high mortality rate frequently due to multiorgan failure.
| Case Report | ▴Top |
A 24-year-old unmarried lady presented herself with complain of continuous high grade fever for 2 months associated with chills and rigors, especially aggravated during evening hours. She also had moderate pain affecting all joints, not associated with swelling, and the pain was not fleeting or migratory in type. Cough started 2 months ago which was dry, and the cough increased at night. She had loss of appetite and weight.
On examination, we noticed that though she was moderately built, she was poorly nourished. She was pale, icteric, had bilateral pitting pedal edema and generalized lymphadenopathy. The enlarged lymph nodes were discrete, non-tender and firm in consistency. She was febrile, tachypneic and had tachycardia, and the blood pressure was 140/80 mm Hg. Abdominal examination showed mild hepatomegaly which was tender and firm, and there was no splenomegaly. Respiratory system examination showed features of right sided moderate pleural effusion. Rest of the systemic examination was unremarkable.
Investigations showed hemoglobin 10 g/dL, total WBC count 7,900/µL, platelet count 0.2 × 109/L and ESR 42 mm in 1 h. Peripheral smear showed normocytic normochromic anemia with evidence of hemolysis and severe thrombocytopenia (Fig. 1). Her corrected reticulocyte count was 5% and the direct and indirect coombs tests were negative. Urine analysis was normal. Her biochemical parameters revealed random blood sugar 75 mg/dL, urea 24 mg/dL, creatinine 0.8 mg/dL, sodium 125 mmol/L, potassium 2.0 mmol/L, AST 122 IU/L, ALT 29 IU/L, alkaline phosphatase 334 IU/L, total bilirubin 3.5 mg/dL, direct bilirubin 2.5 mg/dL, total protein 6.8 g/dL, albumin 2.3 g/dL and globulin 4.5 g/dL. Her prothrombin time and partial thromboplastin time were normal. ECG showed sinus tachycardia and U waves. Chest X-ray showed mild right sided pleural effusion. Arterial blood gas analysis showed fully compensated metabolic acidosis. Abdomen USG showed mild hepatosplenomegaly with small periportal node and mild ascites. Pleural fluid was exudative in nature with lymphocytic pleocytosis. Pleural fluid adenosine deaminase was normal. Pleural fluid acid fast bacilli, gram stain, malignant cytology and acid fast bacilli culture were negative. Based on the history, clinical examination and initial investigations, possibilities of diagnosis that came to us for consideration were infections, hematological malignancies or connective tissue diseases. Etiological workup including dengue, leptospira, Brucella, Rickettsia, EBV, HIV, hepatitis B and C serologies, and antinuclear antibody were negative. Test for malarial parasite was negative and blood cultures did not reveal any growth. Echocardiogram and bone marrow aspirate were also normal.
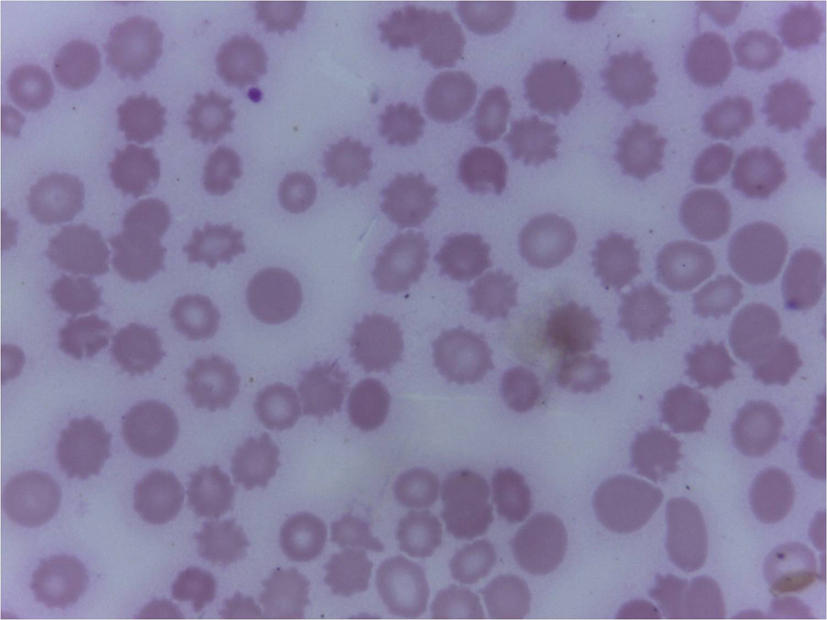 Click for large image | Figure 1. Peripheral smear showing fragmented RBCs and microspherocytes as a part of hemolysis (Leishmans 400 x). |
On the third day of admission, she had fall in oxygen saturation, severe hypotension and two episodes of generalized tonic clonic seizures, and was intubated and mechanically ventilated. Investigations done on that day showed worsening of anemia (hemoglobin 8.5 g/dL) and leucopenia (WBC count 3,900/µL) with a significant fall in ESR to 20 mm in 1 h and a platelet count of 0.2 × 109/L. Her renal function tests also showed worsening. Since the patient had a rapid worsening of her clinical condition, the possibility of development of a complication like thrombotic thrombocytopenic purpura, disseminated intravascular coagulation or HLH was considered. Meanwhile, she was receiving broad spectrum antibiotics, inotropic supports and was mechanically ventilated. Her repeat prothrombin time and partial thromboplastin time were normal. Her serum ferritin was 50,000 µg/L, serum lactate dehydrogenase 10,000 IU/L, serum triglycerides 219 mg/dL and serum fibrinogen 180 mg/dL, all of which propounded HLH. Based on these investigations, the possibility of HLH was considered and started only on steroids, since the diagnosis was not confirmed. But unfortunately, she succumbed to her illness on the fourth day of admission. Later the bone marrow (Fig. 2) and lymph node (Fig. 3) biopsies showed hemophagocytosis. Since most of the etiological workup were negative, an EBV-PCR was performed which was positive.
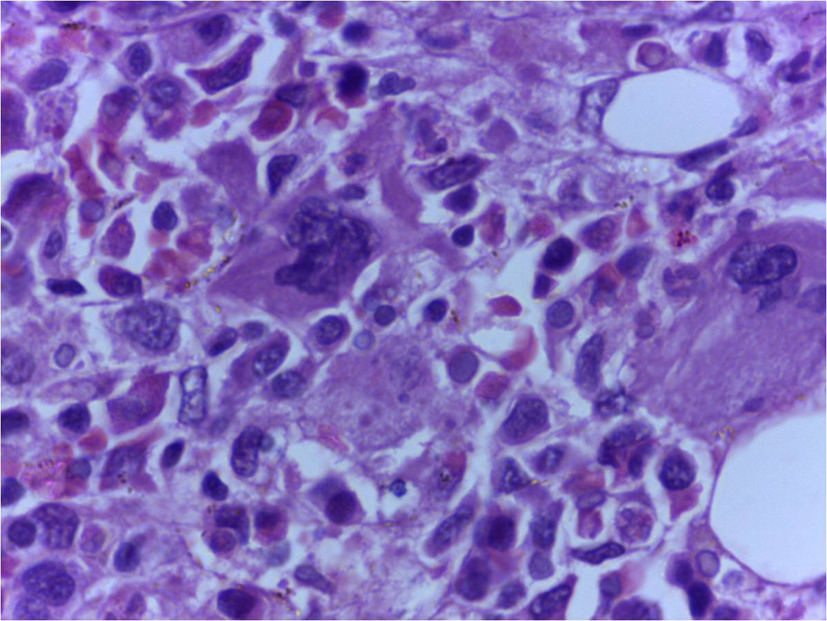 Click for large image | Figure 2. Bone marrow trephine biopsy showing histiocytes with phagocytic debris in cytoplasm (H&E, 400 x). |
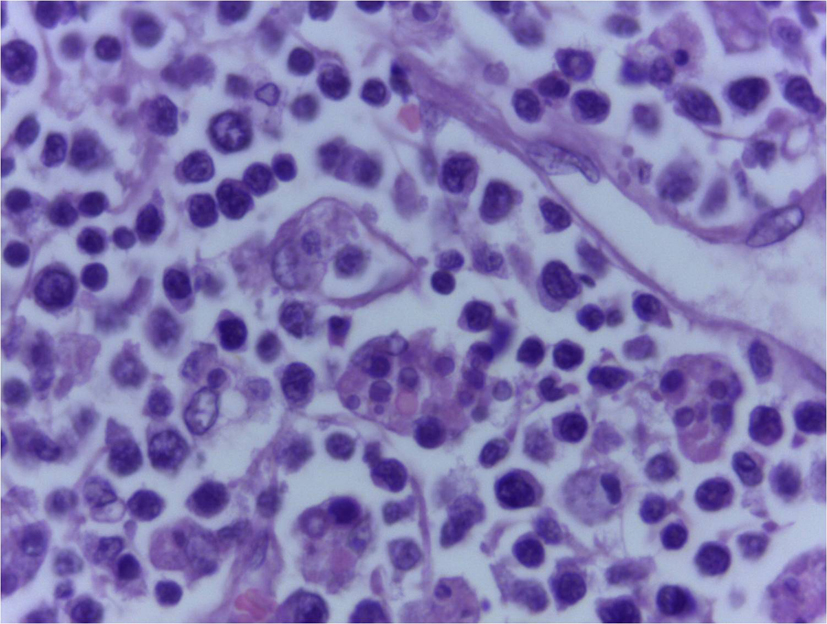 Click for large image | Figure 3. Lymph node biopsy showing engulfed RBCs in cytoplasm of histiocytes (H&E, 400 x). |
| Discussion | ▴Top |
HLH is a frequently fatal and likely underdiagnosed disease involving a final common pathway of hypercytokinemia, which can result in end-organ damage and death. Hemophagocytic syndromes represent a severe hyperinflammatory condition with the cardinal symptoms of prolonged fever, cytopenias, hepatosplenomegaly and hemophagocytosis by activated, morphologically benign macrophages [1]. Hemophagocytic syndrome is caused by a dysregulation in natural killer T-cell function, resulting in activation and proliferation of lymphocytes or histiocytes with uncontrolled hemophagocytosis and cytokine overproduction.
This can be either primary, with a genetic etiology, or secondary, associated with malignancies, autoimmune diseases, or infections. Infections associated with hemophagocytic syndrome are mostly viruses, particularly EBV and other herpes viruses, HIV, influenza, parvovirus, and hepatitis viruses, as well as bacterial, fungal, and parasitic organisms [2]. Diagnosing HLH is challenging and often a strenuous process, because of its various accompanying factors like its rarity, variable presentations, the complexity of diagnostic criteria and concern regarding to the specificity of current diagnostic criteria [3]. HLH presents in many forms: fever of unknown origin (FUO), hepatitis/acute liver failure, sepsis-like, Kawasaki-like and neurologic abnormalities. All of the HLH diagnostic criteria may not be present initially, so it is important to follow clinical signs and laboratory markers of pathologic inflammation repeatedly, and constantly to identify the trends and arrive at the right diagnosis. A high index of suspicion in patients with these recurring symptoms, is essential to have a more quick and accurate diagnosis.
EBV is the most frequent infection associated with HLH. The manifestations of EBV associated HLH varies widely from spontaneously resolving inflammation to incessant disease [4]. EBV-HLH has been associated with acute infections not only in B cells, but also in T cells and NK cells. Most descriptive reports of EBV-HLH have arisen from study centers in East Asia, with the majority focused on primary EBV infection in children or adolescents [5]. Of the rare cases of adult EBV-HLH reported, the majority have been in individuals of East Asian origin [6], although isolated cases arising in adults of other ethnicities have also been recorded.
EBV-HLH has been described in the context of immunosuppression, even though most patients are apparently immunocompetent. The high mortality rate among patients with EBV-HLH is, at least in part, due to delays in diagnosis. The similarities between its initial clinical presentation and that of a wide range of infective and inflammatory conditions, which is often compounded by the apparent absence of hemophagocytosis on initial biopsy delays the diagnosis. Although individual findings in HLH may be mimic features of sepsis and multiorgan dysfunction, close follow-up is needed to define the diagnostic criteria for HLH. The current standard of care consists of a decrescendo course of etoposide and dexamethasone, with or without intrathecal therapy [3]. Reported mortalities in secondary HLH vary from 8-22% in rheumatologic-HLH to 18-24% in EBV-HLH [7].
Our patient, mentioned herein, represents one of the rare cases of adult EBV-HLH which falls under a very fatal category. Our patient had a rapid deterioration of her health, and she succumbed to her illness on the fourth day after admission. We did not receive adequate time for instituting the appropriate therapy, since the possibility of HLH arose in our minds only when the patient’s condition started a drastic and unexpected worsening. Since the patient’s bone marrow and lymph node biopsy reports were not available when the patient deteriorated, which prompted us to consider the possibility of HLH, and immediately serum ferritin, lactate dehydrogenase, triglycerides and fibrinogen were sent, the results of which were suggesting HLH. But unfortunately, we could receive the results of the bone marrow and lymph node biopsies which confirmed our diagnosis, only after the patient’s death. Our patient had very high levels of serum ferritin and lactate dehydrogenase, which may suggest HLH. Allen et al in a study found that serum ferritin levels above 10,000 µg/L appear to be specific and sensitive for HLH. For patients without a significant medical history and a new onset of febrile illness with highly elevated ferritin levels, the diagnosis of HLH should be considered [8]. Our patient presented as FUO, and differentiating HLH from other causes of FUO was challenging for us to head towards the correct diagnosis. In one series of clinical cases , children ultimately diagnosed with HLH presented with deceptive fevers above 102 °F for a median of 19 days (range, 4 - 41 days) [9]. In patients with FUO, cytopenias, highly elevated ferritin (> 3,000 g/dL), or soluble CD25 significantly above age-adjusted normal ranges, generally prompt to pursue a complete HLH diagnostic evaluation early in the course of illness itself [3]. Thus, we conclude, based on the evaluation of the specific clinical case that we have narrated herein, and the way we could strive to come to the correct diagnosis in this case, EBV-HLH is a potentially dangerous syndrome of pathologic immune activation characterized by clinical signs and symptoms of extreme inflammation, which can present itself camouflaged as FUO. We hope that our case report would guide physicians to be aware of this rare aggressive hyperinflammatory syndrome and be on the alert while evaluating FUO patients.
| References | ▴Top |
- Janka GE. Hemophagocytic syndromes. Blood Rev. 2007;21(5):245-253.
doi pubmed - Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7(12):814-822.
doi - Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052.
doi pubmed - Fox CP, Shannon-Lowe C, Gothard P, Kishore B, Neilson J, O'Connor N, Rowe M. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults characterized by high viral genome load within circulating natural killer cells. Clin Infect Dis. 2010;51(1):66-69.
doi pubmed - Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, Yamamoto K, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86(1):58-65.
doi pubmed - Imashuku S, Kuriyama K, Sakai R, Nakao Y, Masuda S, Yasuda N, Kawano F, et al. Treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH) in young adults: a report from the HLH study center. Med Pediatr Oncol. 2003;41(2):103-109.
doi pubmed - Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program. 2011;2011:178-183.
doi pubmed - Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50(6):1227-1235.
doi pubmed - Palazzi DL, McClain KL, Kaplan SL. Hemophagocytic syndrome in children: an important diagnostic consideration in fever of unknown origin. Clin Infect Dis. 2003;36(3):306-312.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.