









| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 3, Number 3, September 2014, pages 80-83
Hypernatremia in Acute Myeloid Leukemia With Trisomy 13: A Case Report and Review of Literature of Diabetes Insipidus in Association With Acute Myeloid Leukemia
Kamyar Nadera, Nicole J. Abelb, d, Peter Georgesa, Chady Abboud-Leona, Barry L. Barnoskic, Roland Schwartingc, Linda Devereuxa, Andres Ferbera
aDepartment of Hematology and Oncology, MD Anderson Cancer Center at Cooper, Camden, NJ, USA
bDepartment of Internal Medicine, Cooper University Hospital, Camden, NJ, USA
cDepartment of Pathology, MD Anderson Cancer Center at Cooper, Camden, NJ, USA
dCorresponding Author: Nicole J. Abel, Department of Internal Medicine, Cooper University Hospital, Camden, NJ, USA
Manuscript accepted for publication February 25, 2014
Short title: Hypernatremia in Acute Myeloid Leukemia
doi: https://doi.org/10.14740/jh125w
| Abstract | ▴Top |
Diabetes insipidus (DI) is a disorder characterized by the inability of the body to conserve water. It occurs either via a central mechanism, where anti-diuretic hormone (ADH) is deficient, or via a nephrogenic mechanism characterized by normal ADH secretion and varying degrees of renal resistance to its water-retaining effects. Central DI is mostly idiopathic, but it can be secondary to trauma, surgery, tumors, infections or other infiltrative processes in the brain. Acute myeloid leukemia (AML) is a rare association with central DI, particularly when there are karyotype abnormalities such as monosomy 7 or inversion of chromosome 3q. Here we report a case of DI associated with AML without these chromosomal abnormalities, but with trisomy 13 in a 19-year-old Haitian female.
Keywords: Acute myeloid leukemia; Central diabetes insipidus; Trisomy 13; Monosomy 7; Inv(3)(q21q26)
| Introduction | ▴Top |
Diabetes insipidus (DI) is a disorder characterized by polyuria and polydipsia and is a result of body’s inability to conserve water properly. The disease is categorized into two forms, nephrogenic and central DI. Nephrogenic is characterized by decreased ability to concentrate urine because of resistance to anti-diuretic hormone (ADH) as a result of inherited conditions or drugs such as lithium. Central DI is characterized by reduced or absent synthesis and secretion of ADH. Central DI is idiopathic in 30-50% of cases, but other cases are associated with damage to hypothalamic-neurohypophyseal region secondary to surgery, trauma, primary or metastatic tumors and infiltrative diseases [1]. Central DI has been reported in association with leukemia, particularly acute myeloid leukemia (AML), chronic myeloid leukemia, myelodysplastic syndrome (MDS) and acute lymphoblastic leukemia [2]. The mechanism of DI in leukemia is not well understood. Hemorrhage, thrombosis, infection or tumor cell infiltration of pituitary or hypothalamus may be responsible factors. However, DI development is not observed in many patients with leukemic infiltration of the hypophyso-hypothalamic region [3, 4].
The first association between DI and AML was described in 1987 and has been reported various times afterwards, particularly preceding initial diagnosis of AML and at times as a manifestation of relapse of disease [5]. Cytogenetic abnormalities such as monosomy 7 and 3q21q26 rearrangements/aberrations have been described in association with DI [6, 7]. Here we report a 19-year-old female without these cytogenetic abnormalities, instead with trisomy 13. Trisomy 13 is a rare, recurring clonal chromosome abnormality in myeloid malignancies. To our knowledge, this is the first associated DI with AML associated with trisomy 13 without monosomy 7 or 3q21q26 abnormalities.
| Case Report | ▴Top |
A 19-year-old Haitian female with no past medical history, who immigrated to the United States in 2011, was hospitalized in June 2013 with complaints of epigastric pain along with fevers and malaise. At presentation she denied any nausea, vomiting, diarrhea, headaches, diplopia, weight loss, night-sweats, or urinary symptoms. She was febrile with temperature of 102 °F. Her laboratory values indicated white blood cell count of 2,000/µL with 20% blasts, hemoglobin of 11 g/dL and platelets of 140,000/µL. All her cultures were negative. Her basic metabolic panel and urine analysis were within normal limits. A bone marrow biopsy showed effaced bone marrow with 90% blasts with no reticulin fiber staining. The blast population on flow cytometry was positive for TdT, CD13, CD15, CD34, CD45 and CD117 and negative for MPO, CD2, CD3, CD7, CD14, CD19, CD64 and CD33. Cytogenetic analysis revealed 47,XX,+13<4>/94,XXXX,?add(5)(q31),+13,+13<12>/98,XXXX,+4,+6,+7,+13,+13,+21<3>/46,XX<1> (Fig. 1). Fms-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD), nucleophosmin-1, or CCAAT/enhancer binding protein alpha mutation was not analyzed.
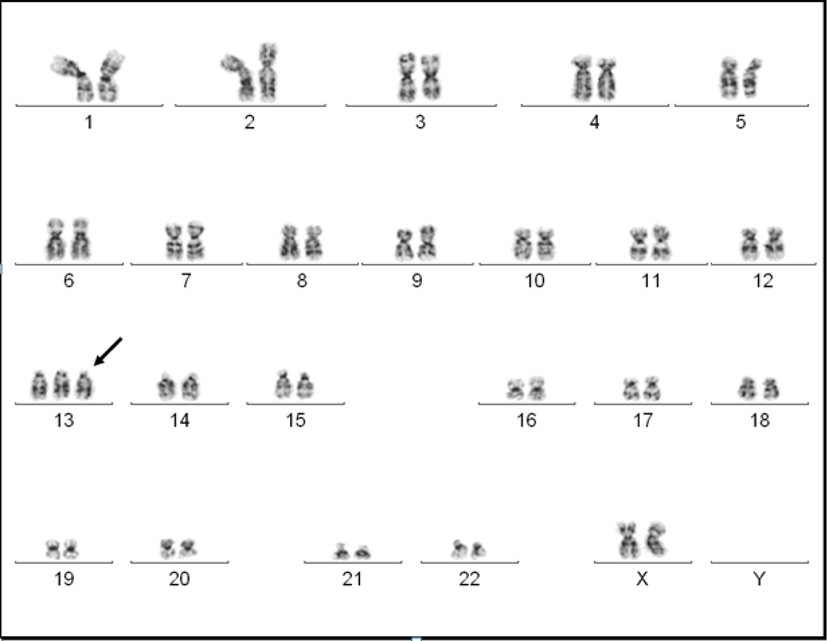 Click for large image | Figure 1. A G-banded karyogram showing trisomy 13. |
After the patient was diagnosed of AML, she began induction chemotherapy with 7 + 3 regimen, which included idarubicin 12 mg/m2 daily for 3 days and cytarabine 100 mg/m2 by continuous infusion for 7 days. Her bone marrow eventually recovered on day 48 with normal hematopoietic elements and trilineage hematopoiesis. After induction her hospital course was complicated by neutropenic fevers, Klebsiella pneumonia bacteremia and Ascaris lumbricoides worm infection which were appropriately treated.
In the second week of treatment the patient manifested increasingly poor oral intake with nausea. On day 16, it was noted that she had elevated serum sodium of 168 mmol/L and measured serum osmolality of 354 mOsm/kg. Her urine osmolality was 155 mOsm/kg. MRI of brain showed absence of normal bright posterior pituatory on T1-weighted imaging suggesting disruption of production of vasopression in the pituicytes and normal pituitary stalk (Fig. 2). She received free water infusion and 2 days of desmopressin. Her serum sodium level normalized to 144 mmol/L on day 24. After further inquiries, patient reported polyuria, nocturia and polydipsia of about 20 bottles of water daily for some months prior to presentation. A water deprivation test was not performed for diagnosis.
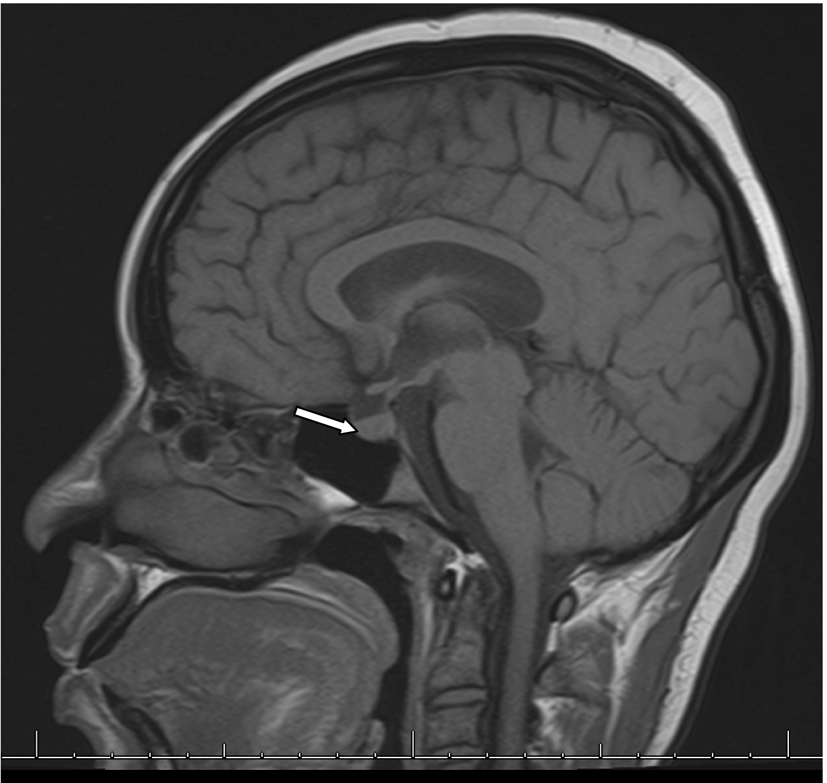 Click for large image | Figure 2. MRI of brain with and without contrast showing absence of bright T1-weighted bright spot in posterior pituitary. |
Patient currently has received consolidation therapy with high-dose cytarabine, 3 g/m2, and currently remains in remission. She does not complain of any polyuria or polydipsia and her sodium remains in normal limits.
| Discussion | ▴Top |
The major symptoms of DI are polyuria and polydipsia. Only when the patient cannot compensate by drinking large amounts of water will hypernatremia develop, as it did for our patient. Her symptoms of thirst presented prior to the typical leukemic picture on presentation but we later suspected her DI was associated with her leukemia based on the results of imaging, laboratory data and clinical scenario, even in the absence of pathological confirmation.
Most of the ADH is synthesized in the supraoptic and paraventricular nuclei and then secreted from the posterior hypophysis. Although DI and AML have been linked, it is a rare association and its mechanism remains not well understood. The earliest proposed mechanism has been leukemic infiltrate, yet this has not been validated. In 1973, Masse et al reported 46% of acute leukemic patients on autopsies had peri-hypophyseal leukemic infiltrates and not all of their karyotypes were known [8]. Kimmel et al reported pituitary involvement in 0.6% of leukemia cases, which is a remarkable discrepancy to the Masse’s 1973 report and makes the true prevalence of leukemia-associated central DI ambiguous. This might be due to the fact that not all leukemic patients with pituitary involvement have clinical DI, as you require near-total destruction of the posterior hypophysis or involvement of about 50% of the fibrils of the hypothalamo-hypophyseal pathway. In addition to the infiltrative process, other possible causes in AML patients include hemorrhage, thrombosis and infection. Given the heterogeneity of observed pathologies with the aforementioned culprits, molecular pathogenesis of DI in AML patients has sparked an interest in the past few decades.
Association with specific chromosomal abnormalities has also been reported in leukemic patients. Monosomy 7 is strongly associated with both MDS and AML, occurring in approximately 5% of de novo cases and 11% secondary cases and has poor prognosis [9]. De la Chapelle and Lahtinen proposed that monosomy 7 predisposes patients with acute leukemia or MDS to DI. This is thought to be due to abnormal expression of neutrophil migration (NM) gene, which maps to chromosome 7 and codes for glycoprotein GP130 on surface of granulocytes, which predisposes patient to DI. NM gene is important for neutrophil chemotaxis and may be related to leukemic infiltration of pituitary gland [10, 11].
Anomalies of chromosome 3, particularly 3q26 breakpoint with inv(3)(q21q26), in AML patients have been associated with DI. It is interesting to note based on reported cases that, sometimes AML patients with latter chromosome abnormality that have normal or high platelet count, and dysmegakaryopoeiesis are deemed to have “3q21q26syndrome”, which has a poor prognosis [12]. Nussey et al described two mechanisms, where ectopic virus integration (EVI-1) site gene, which is located on chromosome 3q, is transcriptionally activated and often associated with thrombocytosis. This may interfere with ADH action because greater than 90% of circulating ADH is bound to platelets, although thrombocytosis is not always present in 3q21q26 syndrome. Other proposed mechanism is via direct hypothalamic neuroendocrine secretion interference [13, 14].
Trisomy 13 is a recurring but rare chromosomal abnormality in AML. Although AML with trisomy 13 is morphologically heterogeneous and has been reported in most of the French-American-British types, a substantial proportion has been classified as M0 or M1 [15-17]. They usually have shown low remission rates and poor response to treatment, possibly due to correlation of trisomy 13 with RUNX1 mutation. RUNX1 mutation has been shown to have increase in FLT3 overexpression due to an unknown mechanism [16]. Based on our literature search, there have been no reported cases of trisomy 13 with DI without aforementioned cytogenetic abnormalities. Harb et al reported two cases of trisomy 13 with DI manifestations; however, they were in concurrence with monosomy 7. Given that the MRI of our patient with trisomy 13 did not show leukemic infiltration but rather impaired production of vasopressin, a molecular etiology is suggested as the culprit, which remains to be elucidated. In conclusion, it is important to realize that central DI can be as one of the first signs of acute leukemia or possibly indicating relapse, even though it is considered a rare complication.
Conflict of Interest
The authors declare no conflict of interests with this report.
| References | ▴Top |
- Makaryus AN, McFarlane SI. Diabetes insipidus: diagnosis and treatment of a complex disease. Cleve Clin J Med. 2006;73(1):65-71.
doi - Yamagami K, Yoshioka K. Central diabetes insipidus presenting as an early sign of acute myeloid leukemia. Clin Adv Hematol Oncol. 2012;10(6):400-401.
pubmed - Kimmel DW, O'Neill BP. Systemic cancer presenting as diabetes insipidus. Clinical and radiographic features of 11 patients with a review of metastatic-induced diabetes insipidus. Cancer. 1983;52(12):2355-2358.
doi - Castagnola C, Morra E, Bernasconi P, Astori C, Santagostino A, Bernasconi C. Acute myeloid leukemia and diabetes insipidus: results in 5 patients. Acta Haematol. 1995;93(1):1-4.
doi pubmed - Ra'anani P, Shpilberg O, Berezin M, Ben-Bassat I. Acute leukemia relapse presenting as central diabetes insipidus. Cancer. 1994;73(9):2312-2316.
doi - de la Chapelle A, Lahtinen R. Monosomy 7 predisposes to diabetes insipidus in leukaemia and myelodysplastic syndrome. Eur J Haematol. 1987;39(5):404-411.
doi - Nussey SS, Ang VT, Bevan DH, Jenkins JS. Human platelet arginine vasopressin. Clin Endocrinol (Oxf). 1986;24(4):427-433.
doi - Masse SR, Wolk RW, Conklin RH. Peripituitary gland involvement in acute leukemia in adults. Arch Pathol. 1973;96(2):141-142.
pubmed - Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92(7):2322-2333.
pubmed - Slater SE, Maccallum PK, Birjandi F, Gibbons B, Lister TA. Acute myelogenous leukemia (AML) and diabetes insipidus (DI): further association with monosomy 7. Hematol Oncol. 1992;10(3-4):221-223.
doi pubmed - Harb A, Tan W, Wilding GE, Battiwalla M, Sait SN, Wang ES, Wetzler M. Acute myeloid leukemia and diabetes insipidus with monosomy 7. Cancer Genet Cytogenet. 2009;190(2):97-100.
doi pubmed - Jotterand Bellomo M, Parlier V, Muhlematter D, Grob JP, Beris P. Three new cases of chromosome 3 rearrangement in bands q21 and q26 with abnormal thrombopoiesis bring further evidence to the existence of a 3q21q26 syndrome. Cancer Genet Cytogenet. 1992;59(2):138-160.
doi - Dy P, Chua P, Kelly J, Liebman S. Central diabetes insipidus in the setting of acute myelogenous leukemia. Am J Kidney Dis. 2012;60(6):998-1001.
doi pubmed - Martinelli G, Ottaviani E, Buonamici S, Isidori A, Borsaru G, Visani G, Piccaluga PP, et al. Association of 3q21q26 syndrome with different RPN1/EVI1 fusion transcripts. Haematologica. 2003;88(11):1221-1228.
pubmed - Klaus M, Haferlach T, Schnittger S, Kern W, Hiddemann W, Schoch C. Cytogenetic profile in de novo acute myeloid leukemia with FAB subtypes M0, M1, and M2: a study based on 652 cases analyzed with morphology, cytogenetics, and fluorescence in situ hybridization. Cancer Genet Cytogenet. 2004;155(1):47-56.
doi pubmed - Silva FP, Lind A, Brouwer-Mandema G, Valk PJ, Giphart-Gassler M. Trisomy 13 correlates with RUNX1 mutation and increased FLT3 expression in AML-M0 patients. Haematologica. 2007;92(8):1123-1126.
doi pubmed - Dicker F, Haferlach C, Kern W, Haferlach T, Schnittger S. Trisomy 13 is strongly associated with AML1/RUNX1 mutations and increased FLT3 expression in acute myeloid leukemia. Blood. 2007;110(4):1308-1316.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.