









| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 2, Number 2, October 2013, pages 85-88
A Case of Fatal Pulmonary Embolism in a Patient With Warm Autoimmune Hemolytic Anemia: An Under-appreciated Thrombotic Risk
Santhosh K. Sadashiva, d, Nihar Patela, Christie Hiltona, Dulabh Mongab, Robert Kaplanc
aDivision of Hematology Oncology, Western Pennsylvania Cancer Institute, 4800 Friendship Ave, Suite 2303NT, Pittsburgh, PA 15224-1791, USA
bDivision of Hematology Oncology, West Penn Allegheny Health System, 320 E North Ave, Pittsburgh, PA 15212, USA
cHemostasis Thrombosis Lab, Western Pennsylvania Cancer Institute, 4800 Friendship Ave, Suite 2303 NT, Pittsburgh, PA 15224-1791, USA
dCorresponding author: Santhosh K. Sadashiv, Western Pennsylvania Cancer Institute, 4800 Friendship Ave, Suite 2303NT, Pittsburgh, PA 15224-1791, USA
Manuscript accepted for publication August 15, 2013
Short title: Fatal Pulmonary Embolism
doi: https://doi.org/10.4021/jh95w
| Abstract | ▴Top |
Autoimmune hemolytic anemia (AIHA) is a rare disorder in which the life span of red blood cells (RBCs) is prematurely decreased by hemolysis by either IgG or IgM auto-antibodies directed against red cell membrane antigens. Many factors are known to trigger this auto-antibody production. Symptoms are non-specific and vary according to the degree of anemia. Although considerable advances have been made in understanding the pathogenesis and securing the diagnosis, progress in management has been slow with no established therapeutic guidelines despite the input of many investigators. Lymphoproliferative disorders (LPDs) and venous thromboembolism (VTE) remain the two most clinically significant associated conditions. VTE is a more serious, acute and often lethal complication of AIHA. There are many mechanisms described for the prothrombotic state in patients with AIHA. Despite the thrombotic implications, risk of VTE is often overlooked and still lacking are standardized risk-stratification criteria and evidence-based thromboprophylactic strategies appropriate for the level of risk. With this case report, our goal is to raise awareness of this frequently unrecognized disorder and its potential for fatal consequences. We also summarize some of the mechanisms involved with the goal of stimulating a clinical discussion aimed at appropriately risk stratifying these patients for robust thromboprophylactic strategies.
Keywords: Warm autoimmune hemolytic anemia; Pulmonary embolism; Lupus anticoagulant
| Introduction | ▴Top |
Although rare in incidence, autoimmune hemolytic anemia (AIHA) is the most common cause of acquired hemolytic anemia. The etiology of AIHA is often idiopathic with a wide variety of triggers leading to development of auto-antibodies directed against red cell (RBC) membrane. The clinical presentations are non-specific and depend on the degree of anemia and underlying comorbid conditions.
The complications of AIHA are not well recognized and can often lead to fatal consequences. Venous thromboembolism (VTE) is an acute complication of AIHA and continues to be under appreciated despite being described as a major cause for fatality. Pathophysiological mechanisms leading to a prothrombotic state are usually multifactorial. Studies have also defined a statistically significant association between presence of lupus anticoagulant (LA) and VTE. Despite the absence of established recommendations for thromboprophylaxis, detection of LA in patients with AIHA may help in risk stratification for appropriate anticoagulation. Our case report highlights this rare complication and its often fatal consequence. A brief discussion on some of the mechanisms involved is an attempt to focus attention on the need for risk-stratification and effective thromboprophylactic strategies for these patients.
| Case Report | ▴Top |
A 62-year-old Caucasian male presented with one-week history of progressive dyspnea, fatigue and jaundice. He had no history of fevers, chest pain, upper respiratory symptoms, or recent medication changes. His vital signs were stable on presentation with oxygen saturation of 94% on 2 L of supplemental oxygen via nasal cannula. Apart from scleral icterus, the remainder of his physical exam was unremarkable. His complete blood count demonstrated hemoglobin of 6 g/dL, MCV of 94.5, leukocyte count of 20.7 K/mcL and platelet count of 149 K/mcL. Further investigation revealed an elevated indirect bilirubin of 4.7 mg/dL, lactate dehydrogenase of 1286 U/L and an undetectable haptoglobin. A direct antiglobin titer against IgG was 4+ and he was diagnosed with warm autoimmune hemolytic anemia (WAIHA). He began treatment with solumedrol at a dose of 1 mg/kg body weight and sequential compression stockings for deep vein thrombosis prophylaxis. After a brief delay in transfusion due to incompatibility, he was transfused with 2 units of packed red blood cells and a post-transfusion hemoglobin was 6.2 g/dL. Over the course of next 48 hours, his dyspnea continued to progress despite increasing his supplemental oxygen. Blood and urine cultures remained negative. Computerized tomography (CT) of his chest, abdomen and pelvis revealed multiple bilateral small pulmonary emboli and a non-specific left upper lobe pleural/parenchymal consolidation. No lymphadenopathy was noted (Fig. 1a, b, c). He subsequently developed sudden bradycardia with immediate progression to pulseless electrical activity. Despite prolonged cardiopulmonary resuscitation and advanced cardiac support, he could not be revived. After discussion with family, an autopsy was requested. Autopsy identified a large pulmonary embolus extending from the right ventricle and atrium into the pulmonary artery as the cause of his death.
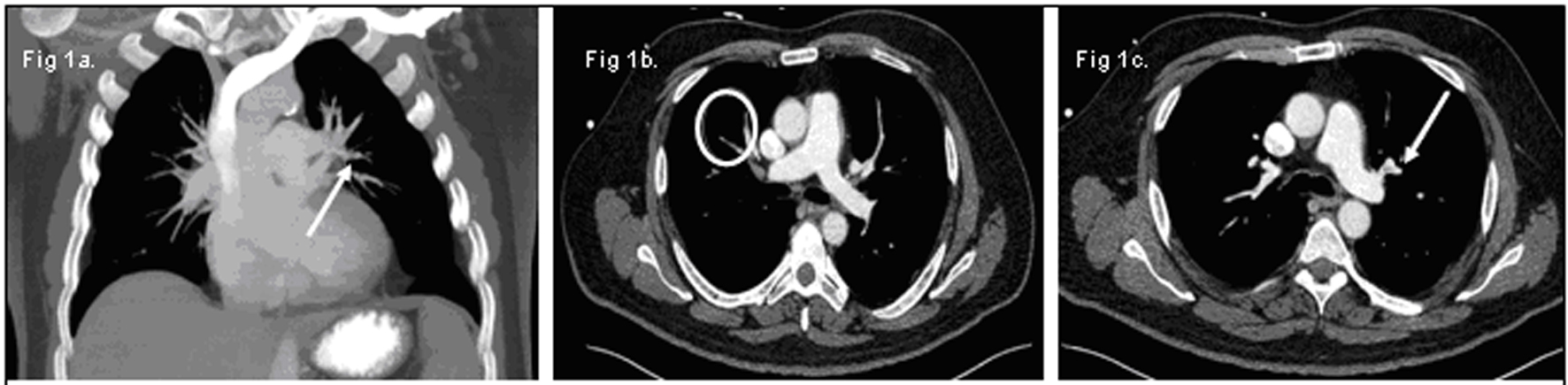 Click for large image | Figure 1. (a) Small pulmonary emboli in the left upper lobe (white arrow). (b) Small pulmonary emboli in right lower lobe (white circle). (c) Small pulmonary emboli in left lower lobe (white arrow). |
| Discussion | ▴Top |
AIHA is a rare disorder characterized by decreased RBC longevity due to anti-RBC antigen IgG/IgM autoantibody-mediated hemolysis. AIHA is the most common cause of acquired hemolytic anemia [1]. There are 2 major forms of AIHA distinguished by the involvement of either IgG (warm antibody) versus IgM (cold antibody) and each subtype has its own unique properties [2]. There are a wide variety of potential processes (viral infection, drugs, autoimmune and connective tissue disorders, malignancy, lymphoproliferative disorders, and so on) which can trigger antibody production [3]. Clinical manifestation depends on the severity of anemia, underlying etiology and associated patient comorbidities. Symptoms of AIHA are non-specific and can range from asymptomatic mild anemia to severe symptomatic anemia requiring urgent treatment. Diagnosis is based on a thorough clinical history and physical examination with laboratory investigation focused on hemolytic parameters and detection and characterization of the culprit antibody [4].
With regard to management of WAIHA, glucocorticoids are the mainstay of treatment and produce a clinically significant response in the majority of patients; however, many patients may need prolonged courses of steroids and become steroid-dependent, while others may require addition of a second agent. Splenectomy and rituximab (monoclonal anti-CD20 antibody) can be regarded as the most effective and well established second-line therapies, but both have limited long term efficacy data [5]. Danazol and other immunosuppressive agents are sometimes employed in refractory cases or after first or second relapses. Transfusion of RBCs in these patients can be challenging due to the presence of red cell alloantibodies in the patient’s plasma, and requires sound clinical judgment and the appropriate blood-bank guidance. In addition, adequate management also encompasses therapy targeting the underlying etiology [5].
Two serious clinical conditions associated with warm AIHA are an underlying lymphoproliferative disease and VTE. LPD may precede (in majority of cases) or follow the diagnosis of AIHA [6, 7]. Advanced age, presence of an underlying autoimmune disease, and the circulation of a monoclonal gammopathy are considered risk factors that may lead to the development of LPD [6, 7].
VTE, the other major complication, is often acute and in some instances may be fatal. This lethal complication has been historically under-appreciated despite having been recognized as early as the 1960’s. A review of 47 patients with AIHA revealed that pulmonary embolism was the most common cause of death [8]. There are several proposed pathophysiologic mechanisms that are believed to contribute to AIHA-induced venous thrombosis. Cytokine-induced expression of tissue factor on monocytes and endothelium [9], hemolysis resulting in exposure of the negatively-charged membrane phospholipid, phosphatidylserine [10] and release of microparticle by-products from RBC fragments during hemolysis are all thought to trigger the coagulation cascade [11]. Other studies have described increased tissue factor expression on endothelial cells and in microparticles [12] and sequestration of nitric oxide by plasma hemoglobin released from the damaged RBCs leading to uninhibited platelet aggregation [13] as other processes responsible for the hypercoagulable state in autoimmune hemolytic disorders. Another investigation found an increased incidence of antiphospholipid antibodies (APLA) in the plasma of patients with AIHA who developed episodes of venous thromboembolism [14]. In the same study, it was also noted that approximately 15% of AIHA patients who developed thrombosis failed to demonstrate evidence of an LA, suggesting the role of other undefined factors [14].
Despite evidence from several studies that support the existence of a prothrombotic state in patients with AIHA, the risk for this complication is often neglected, leading to fatal consequences. Not all patients with AIHA develop venous thromboembolic complications, rendering management of thromboprophylaxis somewhat difficult. Some focus should be devoted to establishing which core factors constitute the true level of thrombotic risk so patients can be risk-stratified to the appropriate level of thromboprophylaxis. One prospective trial conducted in patients with AIHA showed that of the 30 patients in the study, 19 were APLA positive and 8 of the 19 had a documented venous thrombosis translating into a relative risk of 7.5 in AIHA subjects having an LA. This study suggested that detection of the LA in patients with AIHA identifies individuals at significantly increased risk for VTE [14]. Currently some institutions and physicians use LA as a predictive marker to guide their recommendations for thromboprophylaxis; however, formal, evidence-based guidelines for implementing thromboprophylaxis have yet to be established. In an audit of prophylactic anticoagulation in acute exacerbations of severe AIHA, 5 patients out of 15 who did not receive prophylactic anticoagulation had a VTE, and in 3 cases, this resulted in fatal pulmonary embolism. Conversely, only 1 of 21 patients had a venous thromboembolic event when pharmacological prophylaxis was administered [15]. Although this study emphasized both the need and benefits of robust pharmacological prophylaxis, it has not yet been accepted as a routine practice; hence, its use has been limited to only a small number of individual institutions. Physicians caring for these patients may not be sufficiently aware of their substantial VTE risk, with the resulting lack of thromboprophylaxis leaving such patients in a precarious situation. Arterial thrombi appear less common among these patients, but initiating or continuing anti-platelet therapy in the setting of known arterial disease may not be inappropriate.
Several explanations have been cited for the lack of recognition of venous thrombosis as a major complication in these patients. Perhaps, the most plausible is that the clinical presentation of pulmonary embolus is often very similar to that of AIHA itself, with the overlapping symptoms of chest pain, dyspnea and palpitations more often being interpreted as a direct consequence of severe, acute anemia rather than pulmonary emboli. The lack of appreciation in treating physicians of the unique contributing pathophysiologic factors and the associated prothrombotic state likely plays a key role as well.
In this particular case, the discordance between the findings of the CT scan and the autopsy examination could be explained by the scenario of an underlying lower extremity deep vein thrombosis acting as a source of thromboemboli periodically showering the lung, causing progressive dyspnea and the specific chest CT findings. The eventual dislodgement and upward migration of a large embolus would account for the massive fatal pulmonary embolism found at autopsy.
We believe that a high index of clinical suspicion for VTE is warranted in AIHA patients experiencing dyspnea and chest pain despite successful AIHA treatment with a stable or rising hemoglobin level. The use of LA testing may be one reasonable laboratory parameter to be used in AIHA patients to facilitate assigning level of VTE risk and selecting appropriate candidates for robust thromboprophylaxis. Future efforts in clinical investigation should be aimed at identifying the full complement of core clinical and laboratory criteria which will accurately stratify patients with this diagnosis for risk of VTE.
Conflict of Interest
All authors have no conflict of interest.
| References | ▴Top |
- Eaton WW, Rose NR, Kalaydjian A, Pedersen MG, Mortensen PB. Epidemiology of autoimmune diseases in Denmark. J Autoimmun. 2007;29(1):1-9.
doi pubmed - Engelfriet CP, Borne AE, Beckers D, Van Loghem JJ. Autoimmune haemolyticanaemia: serological and immunochemical characteristics of the autoantibodies; mechanisms of cell destruction. SerHaematol. 1974;7(3):328-347.
pubmed - Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24(4-5):143-150.
doi pubmed - Valent P, Lechner K. Diagnosis and treatment of autoimmune haemolyticanaemias in adults: a clinical review. Wien KlinWochenschr. 2008;120(5-6):136-151.
doi pubmed - Klaus L, Ulrich J. How I treat autoimmune hemolytic anemia. Blood. Sept 16, 2010;116(11):1831-1838.
- Genty I, Michel M, Hermine O, Schaeffer A, Godeau B, Rochant H. [Characteristics of autoimmune hemolytic anemia in adults: retrospective analysis of 83 cases]. Rev Med Interne. 2002;23(11):901-909.
doi - Sallah S, Wan JY, Hanrahan LR. Future development of lymphoproliferative disorders in patients with autoimmune hemolytic anemia. Clin Cancer Res. 2001;7(4):791-794.
pubmed - Allgood JW, Chaplin H, Jr. Idiopathic acquired autoimmune hemolytic anemia. A review of forty-seven cases treated from 1955 through 1965. Am J Med. 1967;43(2):254-273.
doi - Hoffman PC. Immune hemolytic anemia—selected topics. Hematology Am SocHematolEduc Program. 2006:13-18.
doi pubmed - Ataga KI, Cappellini MD, Rachmilewitz EA. Beta-thalassaemia and sickle cell anaemia as paradigms of hypercoagulability. Br J Haematol. 2007;139(1):3-13.
doi pubmed - Horne MK, 3rd, Cullinane AM, Merryman PK, Hoddeson EK. The effect of red blood cells on thrombin generation. Br J Haematol. 2006;133(4):403-408.
doi pubmed - Shet AS, Aras O, Gupta K, Hass MJ, Rausch DJ, Saba N, Koopmeiners L, et al. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102(7):2678-2683.
doi pubmed - Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293(13):1653-1662.
doi pubmed - Pullarkat V, Ngo M, Iqbal S, Espina B, Liebman HA. Detection of lupus anticoagulant identifies patients with autoimmune haemolyticanaemia at increased risk for venous thromboembolism. Br J Haematol. 2002;118(4):1166-1169.
doi pubmed - Hendrick AM. Auto-immune haemolyticanaemia—a high-risk disorder for thromboembolism?Hematology. 2003;8(1):53-56.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.