









| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 7, Number 2, May 2018, pages 76-78
A Rare Case of Hermansky-Pudlak Syndrome Type 3
Joel Alcida, b, Jeffrey Kima, David Brunia, Ibiyonu Lawrencea
aDepartment of Medicine, Drexel University College of Medicine, Hahnemann University Hospital, Philadelphia, PA, USA
bCorresponding Author: Joel Alcid, Hahnemann University Hospital, Drexel Medicine, 239 N Broad St, Philadelphia, PA 19102, USA
Manuscript submitted February 22, 2018, accepted March 16, 2018
Short title: Hermansky-Pudlak Syndrome Type 3
doi: https://doi.org/10.14740/jh387w
| Abstract | ▴Top |
Hermansky-Pudlak syndrome (HPS) is a multi-system disorder characterized by oculocutaneous albinism and platelet storage deficiency, which can also lead to prolonged bleeding, pulmonary fibrosis, and granulomatous colitis. Lysosome-related organelle dysfunction is responsible for many of the systemic manifestations, including dense body and melanosome deficiency. This report aims to review a case of HPS type 3 in a male Puerto Rican patient who presented to our clinic.
Keywords: Hermansky-Pudlak syndrome; Lysosome; Platelets; Bleeding; Genetic
| Introduction | ▴Top |
Hermansky-Pudlak syndrome (HPS) is a rare autosomal recessive hemorrhagic disorder primarily due to absence of dense bodies in platelets. HPS is known to affect roughly 1 in 500,000 to 100,000 individuals worldwide with an increased incidence in those of Puerto-Rican heritage. The disorder can be sub-classified into nine classic forms depending on the mutation associated with the disease. The clinical syndrome typically seen with HPS consists of oculocutaneous albinism and bleeding diathesis. Types 1, 2 and 4 are associated with pulmonary fibrosis. Less common features include granulomatous colitis and chronic kidney disease. Lysosome-related organelle dysfunction is responsible for many of the systemic manifestations, including dense body and melanosome deficiency. Unfortunately, the ambiguity of the disease along with the molecular heterogeneity of the different sub-variants makes diagnosis and potential management difficult.
| Case Report | ▴Top |
A 25-year-old male from Northwest Puerto Rico, with past medical history of ocular albinism, presented to the outpatient clinic to establish care. Patient reports that as a child, he experienced episodes of epistaxis and easy bruising, requiring hospitalization. He was worked up for coagulation disorders such as hemophilia, which were negative. He also reports requiring blood transfusions as a child. Patient denied any family history of bleeding disorders. His teenage years and early adulthood years were uneventful as he did not require hospitalizations or transfusions.
In 2014 at the age of 22, he had a dental surgical procedure performed and reported excessive bleeding for 2 days after the procedure. The patient was hospitalized at that time to undergo a thorough evaluation. A diagnosis of HPS was considered, based on the external and ocular features of albinism, visual disturbances, and bleeding diathesis. A blood smear specimen was sent to a special coagulation laboratory at the Mayo Clinic for platelet electron microscopic studies and revealed the platelets had virtually no dense bodies. This finding in conjunction with other platelet EM findings was consistent with a variant of HPS (Figs. 1 and 2).
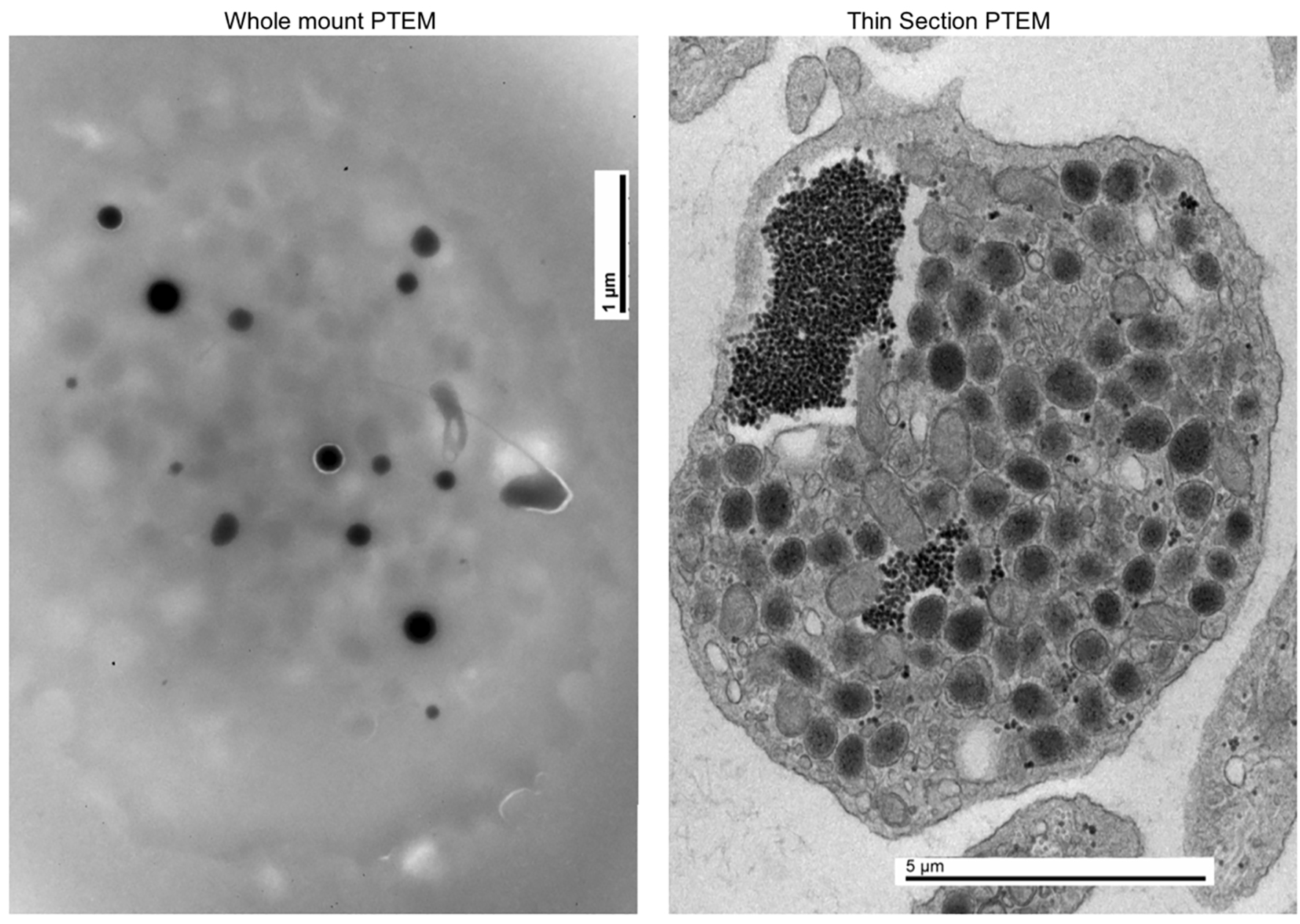 Click for large image | Figure 1. Normal platelet with dense bodies visualized by electron microscopy. |
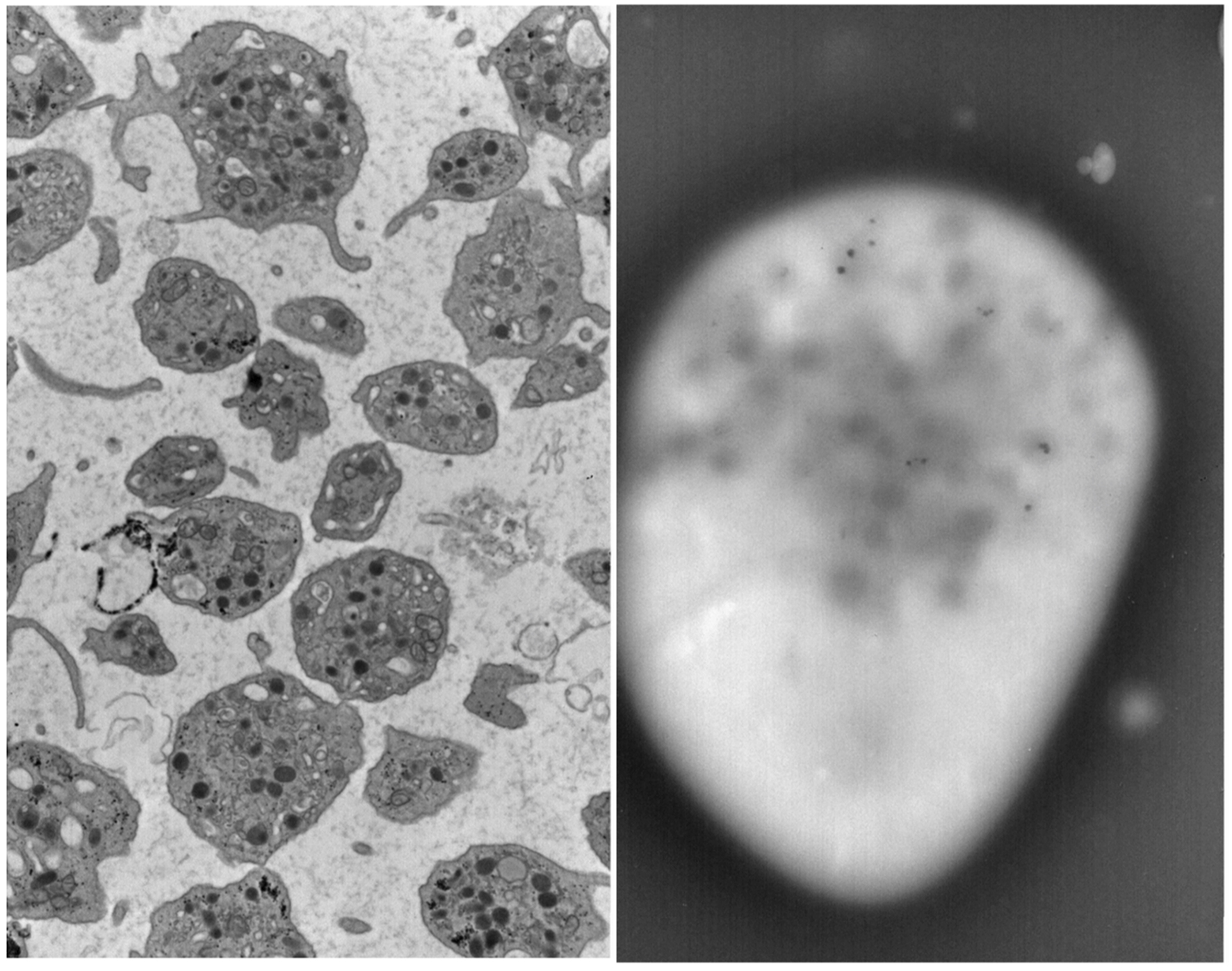 Click for large image | Figure 2. Patient’s platelet with virtually absent dense bodies visualized by electron microscopy. |
He remained stable up until 2016 when he experienced epistaxis and hematemesis. No blood transfusion was required during the hospitalization. He was advised to establish care with a primary care provider and to see a hematologist.
During his initial hematology evaluation, a special genetic test from GeneDx was ordered to possibly confirm the diagnosis of HPS in this patient. The result was positive homozygous for 3.9 kb deletion in the HPS3 gene. The 3.9 kb deletion in the HPS3 gene identified in this analysis could occur in persons of any heritage but is particularly common among individuals of Puerto Rican ancestry who originate for the central region of the island. Homozygosity for this deletion is consistent with the clinical diagnosis for HPS. Genetic counseling was recommended.
At our office, the patient presented with subtle signs of albinism but was otherwise unremarkable. On laboratory analysis, complete blood count showed all cell lines within normal ranges, and platelets specifically were 220,000/µL. We referred him to pulmonology as there is an association with developing pulmonary fibrosis in patients with HPS; however, those with type 3 generally have milder disease. He has not yet had preliminary pulmonary function tests or chest imaging done, but he is currently asymptomatic.
| Discussion | ▴Top |
HPS is a rare hereditary (autosomal recessive) multi-system disorder, characterized by oculocutaneous albinism and platelet storage deficiency, which can also lead to prolonged bleeding, pulmonary fibrosis, and granulomatous colitis. This disorder is most prevalent in the Puerto Rican population, estimated to affect 1 in 500,000 to 1,000,000 individuals worldwide [1, 2]. Mutations in certain genes lead to the clinical manifestations of HPS. There are nine different types of HPS, with type 1 and 4 being the most severe. Mutations in the HPS1 gene cause approximately 75% of the HPS cases from Puerto Rico. Around 45% of affected individuals from other populations have mutations in the HPS1 gene. Other regions where this disorder has been reported include India, Japan, the United Kingdom, and Western Europe. Our patient was homozygous for 3.9 kb deletion in the HPS3 gene, consistent with HPS3, which is also one of the mildest forms [3].
The HPS genes are involved in the formation and trafficking of lysosome-related organelles (LROs), which have been identified in melanocytes, platelets and alveolar type II epithelial cells. Affected individuals can present with oculocutaneous albinism, which is characterized by abnormally light pigmentation of the eyes, skin and hair [4, 5]. Bleeding diathesis is variable, but patients can experience easy bruising, and prolonged bleeding after surgical procedure, and death from hemorrhage has also been reported [6-8].
To further evaluate the patient’s bleeding diathesis thrombocytes were examined closely by microscopic evaluation which revealed a significant decrease in dense granules within the thrombocytes. Dense granules contain factors such as serotonin, calcium, and adenine nucleotides, which are necessary for platelet aggregation, and play a significant role in hemostasis. Studies have shown that affected individuals with HPS can contain up to a 10% reduction in the platelet content of serotonin and ADP [9, 10]. Because of an increased susceptibility to hemorrhage, a hematology consultation is appropriate for patients with HPS prior to any surgical procedure. Prophylactic desmopressin (DDAVP), which augments plasma concentrations of factor VIII, vWF factor, and plasminogen, has been shown to be effective preventing severe bleeding [11]. Aspirin and indomethacin are contraindicated in patients with HPS, because they exacerbate the platelet abnormality. Platelet concentrates should be available in case severe bleeding occurs during or after surgery [12].
Conclusion
This is a unique case of HPS type 3 in a 25-year-old male. As type 3 HPS tends to be more mild, this patient presents an opportunity to follow the course of type 3 HPS in the outpatient medicine setting, while managing the multidisciplinary care he will likely receive for the rest of his life with this disease. Following his pulmonary status, as well as his bowel function, may help elucidate what may trigger these components of HPS in other subtypes and open the possibility for much further investigation.
| References | ▴Top |
- Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, Duffy LF, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome). N Engl J Med. 1998;338(18):1258-1264.
doi pubmed - Dessinioti C, Stratigos AJ, Rigopoulos D, Katsambas AD. A review of genetic disorders of hypopigmentation: lessons learned from the biology of melanocytes. Exp Dermatol. 2009;18(9):741-749.
doi pubmed - Hurford MT, Sebastiano C. Hermansky-pudlak syndrome: report of a case and review of the literature. Int J Clin Exp Pathol. 2008;1(6):550-554.
pubmed - Wei AH, He X, Li W. Hypopigmentation in Hermansky-Pudlak syndrome. J Dermatol. 2013;40(5):325-329.
doi pubmed - Wei AH, Li W. Hermansky-Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res. 2013;26(2):176-192.
doi pubmed - Berber I, Erkurt MA, Kuku I, Kaya E, Koroglu M, Nizam I, Gul M, et al. Hermansky-pudlak syndrome: a case report. Case Rep Hematol. 2014;2014:249195.
doi - Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359-386.
doi pubmed - Witkop CJ, Jr, Quevedo WC, Jr, Fitzpatrick TB, King RA. Albinism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic basis of inherited disease. 6th ed. Vol. 2. New York: McGraw-Hill; 1989. pp. 2905-2947.
doi - White JG. Electron-dense chains and clusters in platelets from patients with storage pool-deficiency disorders. J Thromb Haemost. 2003;1(1):74-79.
doi pubmed - Maurer-Spurej E, Dyker K, Gahl WA, Devine DV. A novel immunocytochemical assay for the detection of serotonin in platelets. Br J Haematol. 2002;116(3):604-611.
doi pubmed - Wijermans PW, van Dorp DB. Hermansky-Pudlak syndrome: correction of bleeding time by 1-desamino-8D-arginine vasopressin. Am J Hematol. 1989;30(3):154-157.
doi - White JG, Witkop CJ. Effects of normal and aspirin platelets on defective secondary aggregation in the Hermansky-Pudlak syndrome. A test for storage pool deficient platelets. Am J Pathol. 1972;68(1):57-66.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.