









| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Case Report
Volume 5, Number 1, March 2016, pages 34-37
Acquired Hemophilia Secondary to Rheumatoid Arthritis: Case Report and Literature Review
Orivaldo Alves Barbosaa, c, Priscila Dourado Evangelistab, Jose Gerardo Araujo Paivab, Tadeu Goncalves de Limaa, Romano Bezerra Brasileirob, Ramiro Rolim Netoa, Cibele Silveira Pinhob, Max Victor Carioca Freitasb
aDepartment of Internal Medicine, Hospital Cesar Cals, Brazil
bDepartment of Rheumatology, Hospital Cesar Cals, Brazil
cCorresponding Author: Orivaldo Alves Barbosa, Department of Internal Medicine, Hospital Cesar Cals, Avenida do Imperador, 545, Centro, Fortaleza, Ceara, Brazil
Manuscript accepted for publication February 24, 2016
Short title: Acquired Hemophilia
doi: http://dx.doi.org/10.14740/jh255w
| Abstract | ▴Top |
Acquired hemophilia is a bleeding disorder caused by the development of autoantibodies against plasma coagulation factors, most frequently against the factor VIII, or type A. We report a case of a 69-year-old patient with rheumatoid arthritis for 6 years with ecchymoses, hematomas and macroscopic hematuria, diagnosed with acquired hemophilia A secondary to rheumatoid arthritis, treated with methylprednisolone, desmopressin, activated prothrombin complex concentrate and cyclophosphamide, with remission.
Keywords: Bleeding; Rheumatoid arthritis; Acquired hemophilia
| Introduction | ▴Top |
Acquired hemophilia A, characterized by the presence of autoantibodies against factor VIII, is a rare condition. This condition is found in patients with autoimmune diseases, hematologic and solid malignancies, postpartum period and pharmaceutical products [1], being the cause of abundant and potentially fatal bleeding.
There are several options for control and treatment of this disease, such as desmopressin, factor VIII concentrate, activated prothrombin complex concentrate (APCC), recombinant human factor VIIa, corticosteroids, cyclophosphamide and rituximab, although to date, there is no gold standard therapy defined. The most used treatment plan, when it is associated with autoimmune diseases, is a combination of methylprednisolone and cyclophosphamide. We describe the case of a female patient with rheumatoid arthritis (RA) who presented with a severe bleeding and was diagnosed with acquired hemophilia A.
| Case Report | ▴Top |
The patient was a 69-year-old woman diagnosed with RA for 6 years (synovitis in small joints of the hands, knee and elbow and positive rheumatoid factor). Four months before the hospital admission, she developed hematoma in her left leg 24 h after minor trauma with macroscopic hematuria and ecchymosis in trunk, upper limbs and face, seeking medical attention without resolution of the condition, being admitted for diagnostic workup and treatment.
On admission, the patient was in compromised state, abdominal pain, bruising in hypogastrium (Fig. 1), dorsal region (Fig. 2) and limbs, melena and secondary osteoarticular changes to RA (fingers buttoning and neck swan). However, the patient had no signs of RA activity, with absence of pain, swelling and heat in affected joints. In the initial tests (Tables 1 and 2), we noticed positive lupus anticoagulant, factor VIII deficiency, presence of inhibitors against factor VIII, anemia, increased activated partial-thromboplastin time (aPTT), hypoalbuminemia, elevated C-reactive protein (CPR), and high ESR.
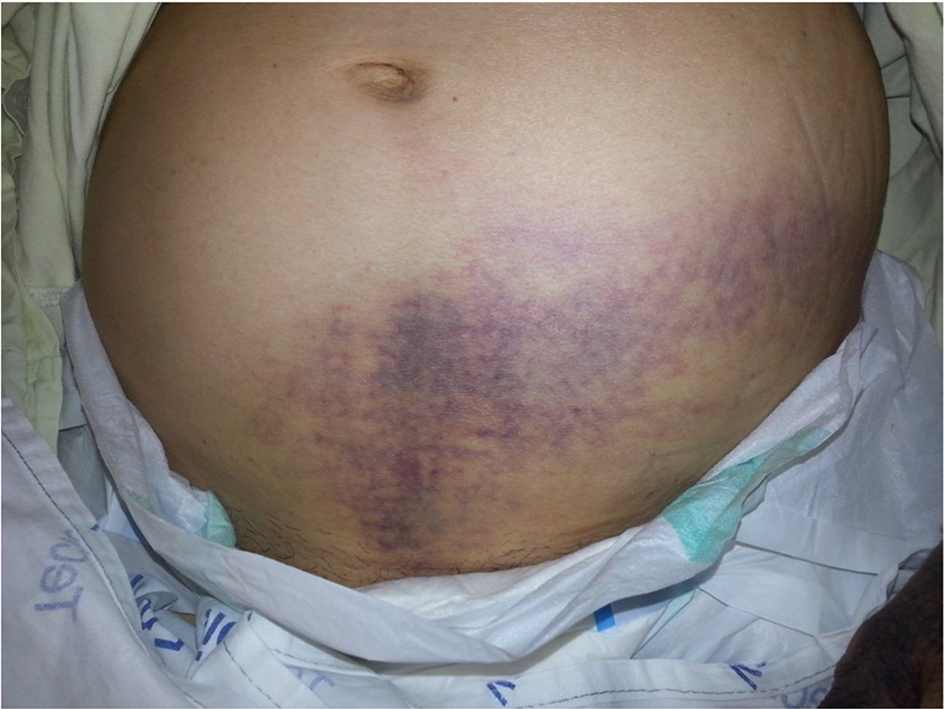 Click for large image | Figure 1. Ecchymosis on the hypogastric region. |
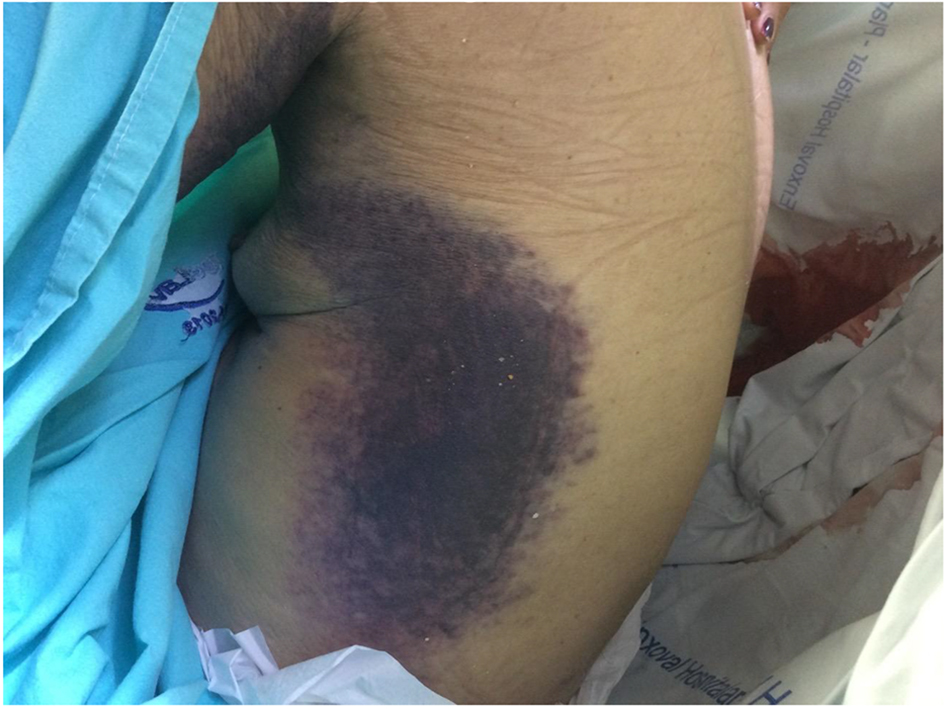 Click for large image | Figure 2. Ecchymosis in dorsal region. |
 Click to view | Table 1. Laboratory Data |
Click to view | Table 2. Serologic Tests |
On the eighth day of hospitalization, the patient progressed with worsening of pain in hypogastric with signs of peritonitis, massive hematochezia, extensive hematoma in the upper limbs after performing arterial puncture, and hemodynamic instability (tachycardia and hypotension). Given the above facts, laboratory tests were performed, which showed new worsening of anemia (Hb: 5.7 g/dL) with maintenance of the enlargement aPTT, and an urgent computerized tomography (CT) which showed amorphous densification in the pelvic cavity, suggestive of hematoma (Fig. 3).
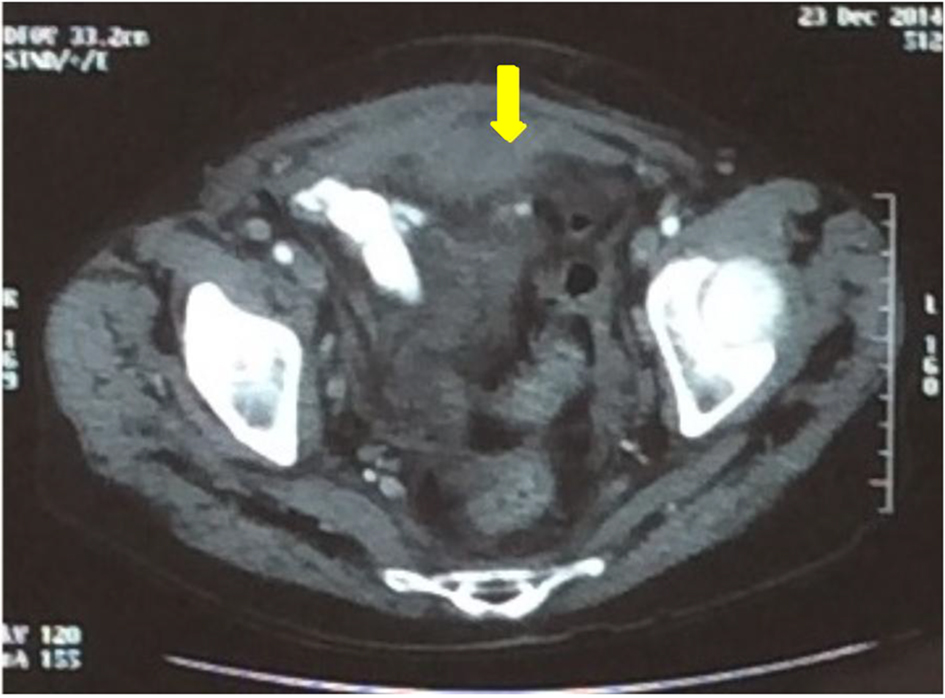 Click for large image | Figure 3. Subaponeurotic hematoma before treatment (arrow). |
With the hypothesis of acquired hemophilia and maintenance of spontaneous hemorrhagic manifestations, further tests were required for laboratory diagnosis (Tables 1 and 2) and it was decided to start therapy with methylprednisolone 1 g for three consecutive days. The patient presented good clinical responses and improved bleeding. After clinical stabilization, survey was conducted for tumors through imaging tests, such as CT of chest, abdomen and pelvis, upper endoscopy, mammography, thyroid ultrasound and colonoscopy, and laboratory tests such as serum protein electrophoresis and also investigation for other immune-mediated diseases that could be responsible for bleeding events.
Twenty days after corticosteroid therapy, patient developed new spontaneous hemorrhagic manifestations (upper limbs and back), severe anemia and new hematoma in the abdominal wall, with factor VIII activity < 1%. The possibility of treatment failure was admitted, requiring the use of APCC and cyclophosphamide, progressing to improvement of hemorrhagic manifestations. The patient improved clinically, with cessation of hemorrhagic manifestations, resolution of abdominal wall hematoma (Fig. 4) and partial correction of the aPTT. She was discharged after 3 months of hospital admission with new factor VIII dosing (3.4%) and programming monthly doses of cyclophosphamide.
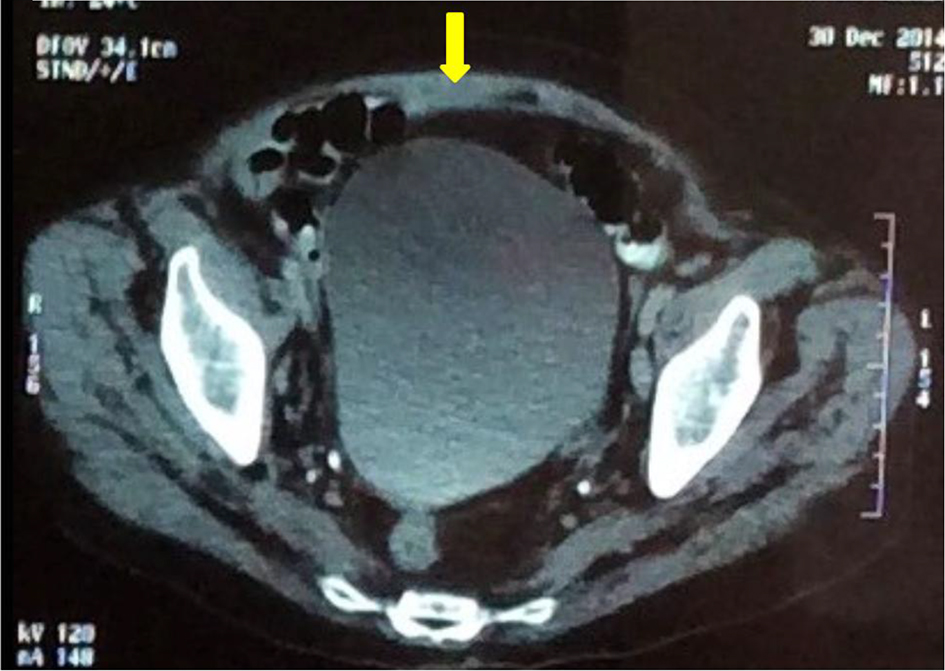 Click for large image | Figure 4. Subaponeurotic hematoma after the treatment (arrow). |
| Discussion | ▴Top |
Acquired hemophilia is a rare bleeding disorder caused by autoantibodies directed against the presence of factor VIII [2]. The factor VIII inhibitors are polyclonal IgG that most commonly are linked to the C2 domains or, less frequently, against the A2 and C3 domain of factor VIII molecule [3].
The incidence of acquired hemophilia A has been reported between 1.34 and 1.48/million inhabitants/year in two recent studies conducted in the United Kingdom [4], probably underestimated because of the difficulty in diagnosis. A series published by Green and Lechner in 1981 studied 215 patients with acquired hemophilia A and concluded that 46% of patients had idiopathic hemophilia, 18% associated with autoimmune diseases, particularly systemic lupus erythematosus and RA, 7% associated with cancer, and the most common dyscrasia of plasma cells and lymphoproliferative disorders; the solid malignancies were found to have a relationship with lung cancer, prostate, colon, pancreas, among others, 5% related to the use of drugs, particularly penicillin, 5% related to dermatological disorders such as psoriasis, exfoliative dermatitis, and erythema multiforme, and 7% related to puerperio [1]. Other diseases are also related to acquired hemophilia A, albeit with a much lower incidence such as inflammatory bowel disease, asthma, COPD, acute hepatitis B and C, multiple transfusions, multiple sclerosis, temporal arteritis, autoimmune hemolytic anemia, autoimmune Goodpasture syndrome, myasthenia gravis, Graves’ disease, autoimmune hypothyroidism, and pemphigus, with a total of 12% of cases [1, 4-6].
Generally, mucocutaneous and soft tissue bleeding is observed in individuals of both sexes, being present in 80% of patients with antibodies against factor VIII. Intramuscular severe bleeding, hematuria, epistaxis, GIT bleeding and intracerebral bleeding are also common clinical manifestations, which are usually severe, requiring successive red blood cell transfusions. The hemarthrosis, a typical manifestation of congenital hemophilia, may also occur, but it is an unusual complaint, occurring in approximately 4.9% of the cases. In selected cases, only anemia can be evidenced due to occult bleeding.
A study of 65 patients showed that 53% had multiple ecchymosis and 42% had hematomas, 37% in soft tissues and 5% in the retroperitoneum. A proportion of patients with retroperitoneal bleeding had a complication neuropathy, femoral nerve compression hematoma; 28% of patients had bleeding of mucous membranes, with 19% TGI related, 8% with the urinary tract and intracranial 1%; 14% of patients had bleeding manifested for a prolonged postoperative period and 2% had hemarthrosis. Life-threatening bleeding is common and is seen in about 9-22% of patients, while mild bleeding that has no therapeutic intervention indication is seen in approximately 30% of cases. Occasionally patients are diagnosed by routine tests without clinical manifestations, and it is known that the spontaneous resolution of the inhibitor occurs in 25% of cases.
Our patient had a prolonged aPPT with a normal prothrombin time and a normal platelet count. A prolonged aPPT with a normal prothrombin time may be due to a deficiency or inhibition of any of the factors that are unique to the intrinsic pathway of the coagulation cascade, namely, factors VIII, IX, and XI [7]. Since this patient had an acquired disorder, a coagulation-factor inhibitor was much more likely to be responsible than a factor deficiency.
To distinguish between the presence of an inhibitor and a deficiency state, coagulation studies should be performed with a 1:1 mixture of the patient’s plasma and normal plasma. Normalization of the aPPT is consistent with a factor deficiency, whereas persistent prolongation of the aPPT indicates the presence of a factor inhibitor.
The decision on the initial treatment should always be based on the severity of bleeding, and the title of the inhibitor. The options used for treatment and control of bleeding include the use of desmopressin, factor VIII concentrates, APCC and recombinant human factor VIIa (rFVIIa) [8-11]. A retrospective analysis by Hay et al has demonstrated a 100% response in patients with acquired hemophilia when rFVIIa was used as first-line treatment, and a response of approximately 75% when it was used in a late phase of the disease [12].
A European study of 331 patients with acquired hemophilia demonstrated the effectiveness of three treatments as initial scheme: glucocorticoids; glucocorticoids plus cyclophosphamide; and glucocorticoids plus rituximab [13]. It was possible to note a higher percentage of responses when glucocorticoids were used as initial therapy associated with cyclophosphamide, requiring shorter treatment time to achieve complete remission of symptoms in the first two treatments compared to the third.
Rituximab has emerged as a promising agent for the eradication inhibitors in patients with acquired hemophilia A with response accounting for 80-100% of cases [14, 15].
Conclusion
Acquired hemophilia, despite being an uncommon disease, should always be suspected in patients with major bleeding associated with the enlargement of aPPT, without alteration of prothrombin time and platelet count. It is worth noting that although the acquired hemophilia is most commonly found in association with autoimmune diseases, cancer and postpartum, because it is a disease with high morbidity and mortality, early diagnosis with an effective therapy is essential for a good prognosis.
| References | ▴Top |
- Cabral A, Romao T, Monteiro JA, Uva LS. Hemofilia Adquirida, a proposito de um caso clinic. Medicina Interna. 1998; 5(1).
- Franchini M, Targher G, Montagnana M, Lippi G. Laboratory, clinical and therapeutic aspects of acquired hemophilia A. Clin Chim Acta. 2008;395(1-2):14-18.
doi pubmed - Reeves BN, Key NS. Acquired hemophilia in malignancy. Thromb Res. 2012;129(Suppl 1):S66-68.
doi - Mahendra A, Padiolleau-Lefevre S, Kaveri SV, Lacroix-Desmazes S. Do proteolytic antibodies complete the panoply of the autoimmune response in acquired haemophilia A? Br J Haematol. 2012;156(1):3-12.
doi pubmed - Grethlein SJ, Besa EC. Acquired Hemophilia. Medscape. 2014.
pubmed - Sborov DW, Rodgers GM. Acquired hemophilia a: a current review of autoantibody disease. Clin Adv Hematol Oncol. 2012;10(1):19-27.
pubmed - Giangrande P. Acquired Hemophilia, Revised edition, World Federation of Hemophilia; Oxford Haemophilia and Thrombosis Centre Oxford, U.K. 2012;38.
- Franchini M, Gandini G, Di Paolantonio T, Mariani G. Acquired hemophilia A: a concise review. Am J Hematol. 2005;80(1):55-63.
doi pubmed - Hay CR, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors Organisation. Br J Haematol. 2006;133(6):591-605.
doi pubmed - Collins PW. Treatment of acquired hemophilia A. J Thromb Haemost. 2007;5(5):893-900
doi pubmed - Baudo F, Collins P, Huth-Kuhne A, Levesque H, Marco P, Nemes L, Pellegrini F, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood. 2012;120(1):39-46.
doi pubmed - Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost. 1997;78(6):1463-1467.
pubmed - Collins P, Baudo F, Knoebl P, Levesque H, Nemes L, Pellegrini F, Marco P, et al. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. 2012;120(1):47-55.
doi pubmed - Wiestner A, Cho HJ, Asch AS, Michelis MA, Zeller JA, Peerschke EI, Weksler BB, et al. Rituximab in the treatment of acquired factor VIII inhibitors. Blood. 2002;100(9):3426-3428.
doi pubmed - Onitilo AA, Skorupa A, Lal A, Ronish E, Mercier RJ, Islam R, Lazarchick J. Rituximab in the treatment of acquired factor VIII inhibitors. Thromb Haemost. 2006;96(1):84-87.
doi
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.