









| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website http://www.thejh.org |
Original Article
Volume 3, Number 3, September 2014, pages 65-71
High-Resolution Analysis of Chromosomal Alterations in Adult Acute Lymphoblastic Leukemia
Lam Kah Yuena, c, Zakaria Zubaidaha, Ivyna Bong Pau Nia, Megat Baharuddin Puteri Jamilatul Noora, Esa Ezaliaa, Chin Yuet Menga, Ong Tee Chuanb, Vegappan Subramanianb, Chang Kiang Mengb
aHematology Unit, Cancer Research Centre, Institute for Medical Research, Kuala Lumpur, Malaysia
bHematology Department, Ampang Hospital, Kuala Lumpur, Malaysia
cCorresponding Author: Lam Kah Yuen, Hematology Unit, Cancer Research Centre, Institute for Medical Research, Jalan Pahang, 50588 Kuala Lumpur, Malaysia
Manuscript accepted for publication May 9, 2014
Short title: Chromosomal Alterations in ALL
doi: https://doi.org/10.14740/jh140w
| Abstract | ▴Top |
Background: Chromosomal alterations occur frequently in acute lymphoblastic leukemia (ALL), affecting either the chromosome number or structural changes. These alterations can lead to inactivation of tumor suppressor genes and/or activation of oncogenes. The objective of this study was to identify recurrent and/or novel chromosomal alterations in adult ALL using single nucleotide polymorphism (SNP) array analysis.
Methods: We studied 41 cases of adult ALL compared with healthy normal controls using SNP array.
Results: Our analysis revealed 43 copy number variant regions, of which 44% were amplifications and 56% were deletions. The most common amplifications were on chromosome regions 8p23.1 (71%), 1q44 (66%), 1q23.3 (54%), 11q23.3 (54%), 12p13.33 (54%) and 8q24.21 (51%). On the other hand, the most common deletions were identified on chromosomes 1p31.1 (76%), 3q26.1 (68%), 11p11.12 (63%), 4q12 (59%), 19p13.2 (59%), 7q11.21 (56%), Xp22.33 (54%), 7q11.21 (51%) and 19p13.11 (51%).
Conclusions: Amplification of chromosome 8p23.1 and deletion of chromosome 1p31.1 were the most frequently found alterations in this study. These chromosomal regions contain genes such as SPAG11B, DEFB104A, DEFB105A, DEFB107A, DEFB106A and SPAG11A. These potential genes may contribute to leukemogenesis in adult ALL. The cytogenetic and molecular mechanisms underlying these chromosome changes deserve further investigations.
Keywords: Acute lymphoblastic leukemia; Adult; Single nucleotide polymorphism array
| Introduction | ▴Top |
Acute lymphoblastic leukemia (ALL) is a heterogeneous disease, resulting from the accumulation of chromosomal alterations either in the form of numerical or structural changes such as amplification, deletion, inversion or translocation. The frequency of chromosomal alterations in adult ALL is 64-85% [1], compared to 60-69% in childhood ALL. Translocation t(9.22), one of the most common recurring chromosomal alterations, is found in 20-40% adult ALL patients [2], and its incidence increases with age. Some of the chromosomal alterations are significantly associated with remission duration, complete remission rate and disease-free survival [1]. Increasing age is also associated with lower remission rates, shorter remissions and poor outcomes in adult ALL [1].
Microarray platforms have been widely used to identify chromosome abnormalities and for gene expression profiling in cancers. These new technologies have increased our understanding of certain nonrandom chromosomal alterations related to morphologic subtypes of acute leukemia [3]. In addition, they provide diagnostic and prognostic information with a direct impact on patient management [4].
Recently, high-resolution single nucleotide polymorphism (SNP) array has been successfully used for screening whole cancer genome. This SNP array contains 1.8 million genetic markers and enables detection of copy number change, uniparental disomy and SNP. In this preliminary study, we analyzed a total of 41 adult ALL samples using the SNP array to detect large and small chromosome changes as well as novel abnormality regions, which may contribute to proliferation and progression of adult ALL.
| Materials and Methods | ▴Top |
ALL samples and DNA preparation
A total of 41 adult patients diagnosed with ALL between 2005 and 2009 were selected for this study. All ALL cell suspensions were obtained from Cancer Research Centre, Institute for Medical Research. Twenty-one healthy volunteer blood donors were used as normal controls. This study was approved by Medical Research & Ethics Committee. Genomic DNA (gDNA) was extracted from cell suspension using the QIAamp DNA Blood Mini Kit according to the manufacturer’s instructions (Qiagen Inc., Valencia, CA, USA). The concentration and the quality of the DNA were determined using an ND-1000 spectrophotometer (NanoDrop, Thermo Scientific).
SNP array and data analysis
SNP genotyping array experiment was performed according to the Affymetrix SNP 6.0 array standard protocol (Affymetrix Inc., Santa Clara, CA, USA). Briefly, 500 ng of gDNA was digested with NspI and StyI restriction enzymes, and then ligated to the adaptors followed by PCR amplification using TITANIUM™ DNA Amplification Kit (Clontech Laboratories, Montain View, CA, USA). PCR products were purified with Agencourt AMPureVR Magnetic Beads (Agencourt Bioscience Corporation, Beverly, MA, USA) and the amplicons were quantified using an ND-1000 spectrophotometer. Subsequently, the amplicons were fragmented, labeled and hybridized to a Genechip Affymetrix SNP 6.0 arrays at 50 °C for 16 - 18 h in a GeneChipVR Hybridization Oven 640 (Affymetrix Inc.). After washing and staining in a GeneChipVR Fluidics Station 450 (Affymetrix Inc.), the arrays were scanned with a GeneChipVR Scanner 3000 7G (Affymetrix Inc.). The analysis of raw data microarray CEL files was performed using Genotyping Console™ (GTC) version 3.0.1.
Validation of SNP array by MS-MLPA
MS-MLPA kit ME002 (MRC-Holland, Amsterdam, The Netherlands) was used to confirm the alteration regions previously identified by SNP array. Briefly, 100 ng of gDNA in volume of 5 µL was denatured at 98 °C for 5 min and then hybridized for 16 h at 60 °C after adding master mixture. Digestion and ligation were performed at the same time at 48 °C for 30 min. Subsequently, PCR was performed for 33 cycles (30 s at 95 °C, 30 s at 60 °C and 1 min at 72 °C) and PCR products are then separated using capillary sequencer ABI model 3730 (Applied Biosystems Inc., CA, USA). The peak height and area values were obtained by GeneMapper 4.0 analysis software (Applied Biosystems Inc.) and exported to an excel spreadsheet for further processing. Normalization of data and calculation of dosage ratios was performed as described at http://www.mlpa.com/WebForms/WebFormMain.aspx?Tag=wl2zCji\rCGANQgZPuTixouFcfv 0KBwygY5OYiCdPw1kHhw\T3x9R7S3MNJOh3N7. The ratio of normal range was defined in between 0.6 to 1.4. Ratio greater than 1.4 and less than 0.6 were defined as amplification and deletion, respectively.
| Results | ▴Top |
We evaluated chromosomal alterations in 41 adult ALL and 21 normal control samples using SNP array. All samples analyzed could detect at least one or more chromosomal alterations. A summary of chromosomal alterations found in the 41 adult ALL samples is depicted in Supplementary 1 (www.jh.elmerpress.com). A total of 935 chromosomal alteration regions were identified in this study. Forty-three chromosomal alteration regions showed more than 40% alterations, of which 19 (44%) regions were amplifications and 24 (56%) regions were deletions. Out of the 935 chromosomal alteration regions identified, 931 involved microdeletion or microamplification of at least 1 Mb in size, whereas four regions involved alteration exceeding 1 Mb. The mean number of chromosomal alterations was 22.80 per case. The mean number of amplifications was 9.39 and the mean number of deletions was 13.41.
In this study, the most common amplifications were identified on chromosome 8p23.1 (71%), 1q44 (66%), 1q23.3 (54%), 11q23.3 (54%), 12p13.33 (54%), 8q24.21 (51%), 15q11.2 (49%), 17q21.31 (49%), 1p13.3 (46%), 12p13.33 (46%), 12q13.13 (46%), 14q11.2 (46%), Xq21.31 (46%), 8q24.3 (44%), Xq21.31 (44%), 6p25.3 (42%), 7q36.3 (42%), 8p21.3 (42%) and 20q11.23 (42%). The most common deletions were identified on chromosome 1p31.1(76%), 3q26.1 (68%), 11p11.12 (63%), 4q12 (59%), 19p13.2 (59%), 7q11.21 (56%), Xp22.33 (54%), 7q11.21 (51%), 19p13.11 (51%), 4q13.2 (49%), 19p12 (49%), 6p23 (46%), 7q22.3 (46%), 9p23 (46%), 1q25.1 (44%), 3p21.31 (44%), 3q23 (44%), 4q35.2 (44%), 8p21.3 (44%), 14q32.33 (44%), 9p11.2 (42%), 11p11.12-p11.2 (42%), 15q15.1 (42%) and 19p13.2 (42%). Homozygous amplifications on chromosomes 12p13.33 and 17q21.31 were observed in six cases and homozygous amplifications on chromosomes 1q44 and 8p23.1 were observed in four cases. We detected homozygous deletion on chromosome 14q32.33 in three cases. Figure 1 shows the most common alterations at chromosomes 1p31.1 and 8p23.1.
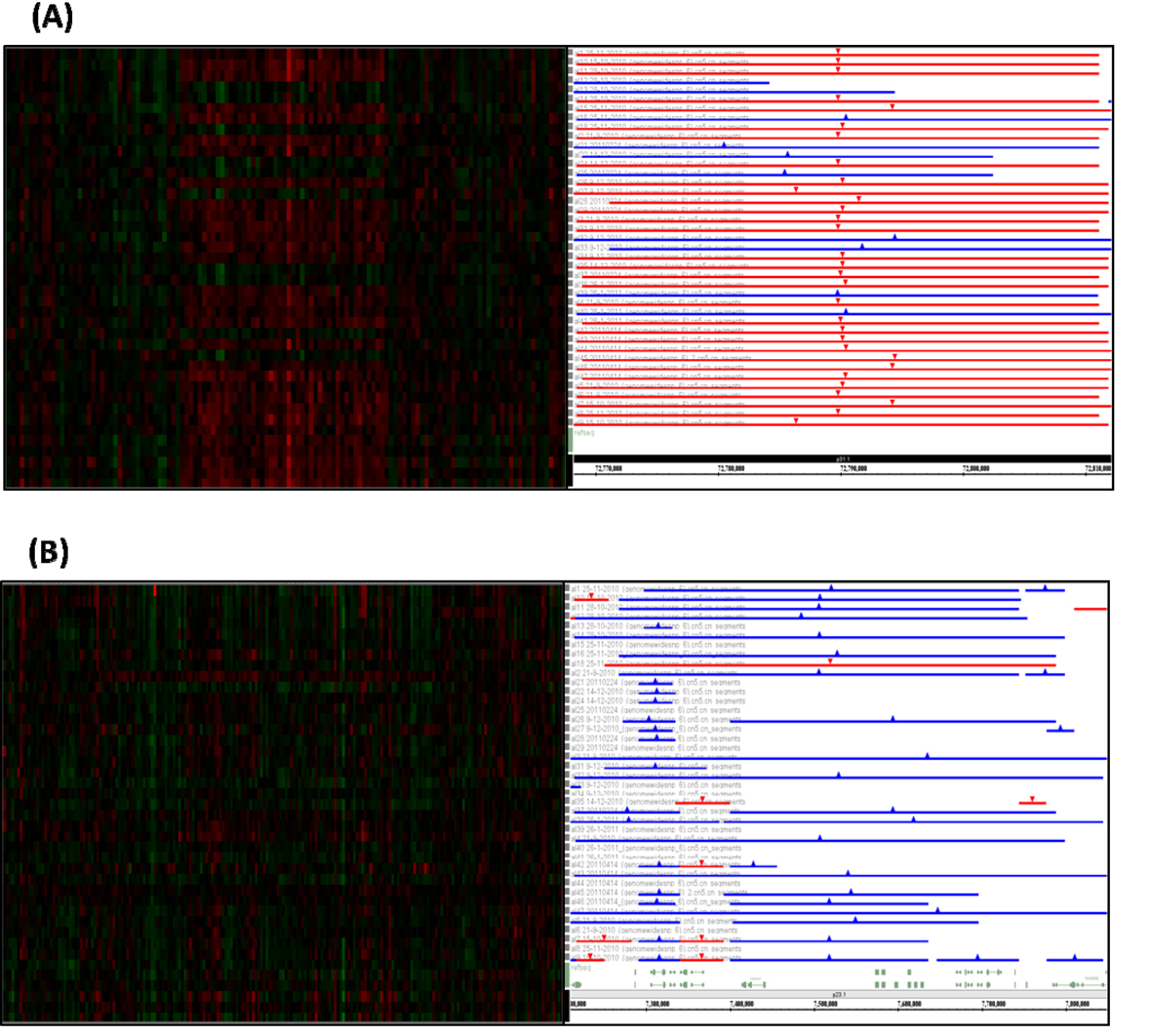 Click for large image | Figure 1. The most common chromosomal alterations in the 41 adult ALL cases. The left lanes are heatmap of Affymetrix SNP array. Red is deletion and green is amplification. The right lanes are segmentation of DNA copy number. Red is deletion and blue is amplification. The most common deletion was found at chromosome chr1p31.1 (A) and amplification was found at chromosome chr8p23.1 (B). |
For validation, MLPA technique was used to confirm that some of the tumor suppressor genes were located within the chromosomal alteration regions as identified by SNP array. The MLPA results were in concordance with the SNP array results (Fig. 2).
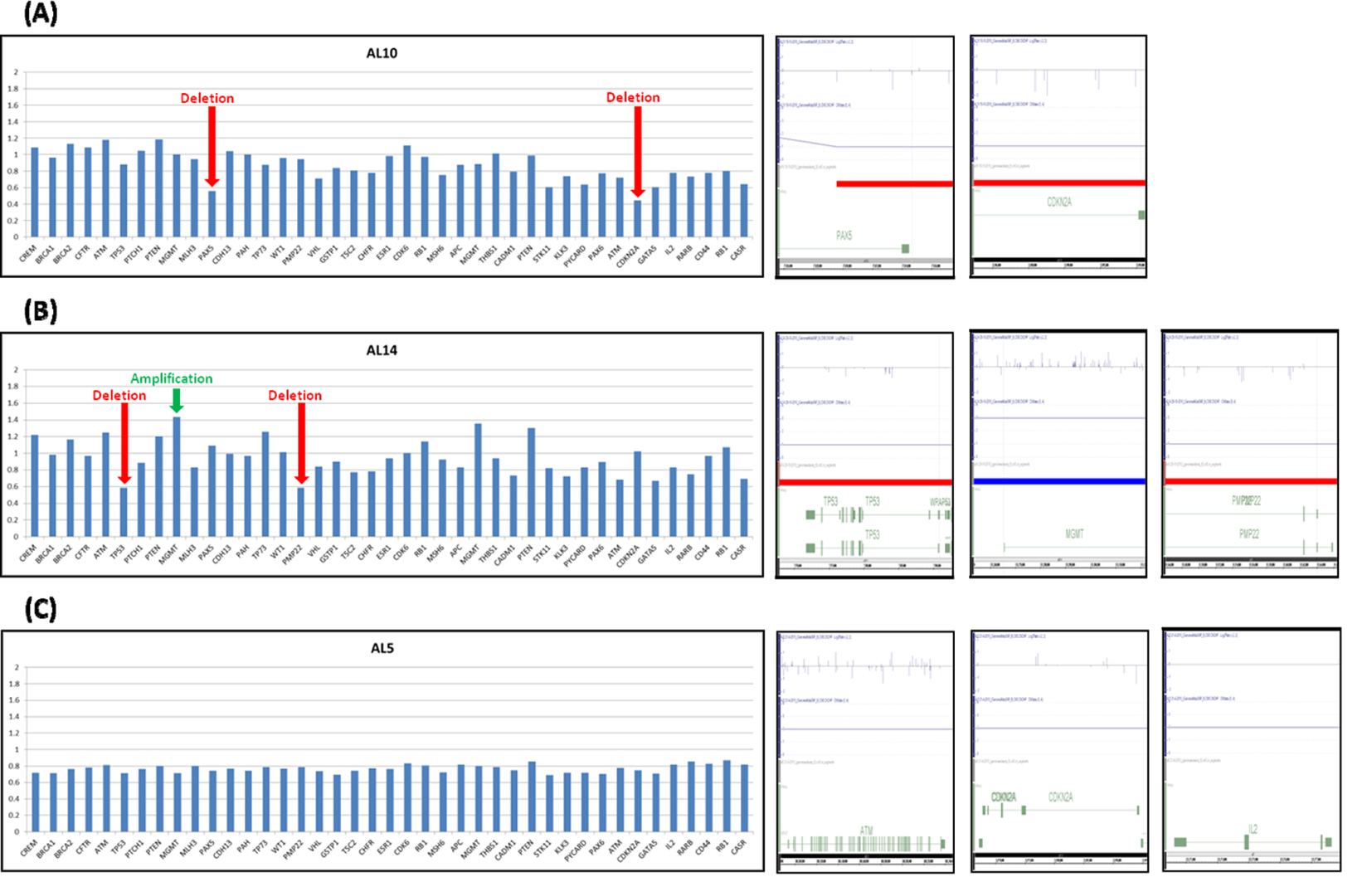 Click for large image | Figure 2. Validation of chromosomal alteration in adult ALL using MLPA technique. The validation results for samples AL10, AL14 and AL5 are labeled as A, B and C respectively. The left lanes showed MLPA results. Ratio < 0.60 was considered as deletion, > 1.40 was considered as amplification. The right lanes showed SNP array results. (A, B) Red arrow indicated that deletion occurred in PAX5 and CDKN2A genes in AL10 sample and TP53 and PMP22 in AL14 sample. (B) Green arrow indicated MGMT gene amplified in AL14 sample. (C) No amplification or deletion was found in AL5 sample by MLPA technique. The results of SNP array were concordance with MLPA result. |
| Discussion | ▴Top |
Chromosome translocations are the most common types of chromosomal abnormalities found in ALL, leading to the generation of fusion genes. Although many of the chromosome translocations were identified by conventional cytogenetics, this method is limited in its ability to detect microamplifications and microdeletions. High density resolution SNP arrays have been applied in hematological malignancies to identify submicroscopic chromosomal alterations such as deletion, amplification, copy-neutral loss of heterozygosity and SNP [5, 6]. SNP arrays contain approximately 1.8 million markers, including 946,000 probes for the detection of copy number variants and 906,600 SNPs.
In this study, we used the high-resolution of SNP arrays to investigate chromosomal alterations in a total of 41 cases of adult ALL, which may not be detected via conventional cytogenetic method. We found that the most common chromosomal alteration was deletion compared to amplification. All cases showed at least one or more chromosomal alterations. A total of 935 chromosomal alteration regions were identified in this study. Some of these chromosomal alteration regions may contain oncogenes, tumor suppressor genes and cancer-related genes, suggesting that these chromosomal alteration regions may play an important role in leukemogenesis.
The most common chromosomal alteration in our study was deletion at chromosome 1p31.1 as observed in 31 out of 41 ALL samples (76%). Previous studies have revealed that deletion of chromosome 1p31-32 is significantly associated with shorter survival in myeloma [7]. Deletions of chromosome 1p31.1 have been detected in many tumor types, including ovarian cancer, breast cancer and lung cancer. The second most common deletion was on chromosome 3q26.1 (68%). Both of these regions are novel findings in this study. Both of these regions have not been identified any tumor suppressor genes or cancer-related genes, suggesting that these regions may contain tumor suppressor genes and cancer-related genes involved in leukemogenesis.
Amplification of chromosome 8p23.1 was observed in 21 out of the 41 (51%) adult ALL samples. This alteration region contains beta-defensin genes such as DEFB103A, DEFB104A, DEFB104B, DEFB105A, DEFB105B, DEFB106A, DEFB106B, DEFB107A, DEFB107B, DEFB109P1B and DEFB4A, and miscRNA genes such as FAM66B, FAM66E, FAM90A10, FAM90A13, FAM90A14, FAM90A18, FAM90A19 and FAM90A7. Defensin gene plays an important role in the innate immune system and expresses short cationic peptides in epithelia and leukocytes [7]. The second most frequent amplification was found at chromosome 1q44 (66%), whereas four cases showed a homozygous amplification at this chromosome region. More recent studies have revealed that amplification of chromosome 1q31.3-q44 is associated with the aggressive NK-cell leukemia group [8] and T-cell leukemia (> 20%) [9]. The results of this study were entirely consistent with Nakashima’s [10] study, of which around 50% samples showed amplifications of chromosome 8p23.1 in leukemia.
In this study, 7-20% cases showed deletion at chromosome 9p21-p22. Some investigators have confirmed that 9p alterations are more likely to occur in ALL compared to other hematological malignancies. The frequency of chromosome 9p abnormality has been reported in 7-13% with not much difference in childhood and adult ALL [11-13]. A previous study has revealed that deletion of chromosome 9p21-p22 is associated with poor outcome in childhood ALL [14]. According to Nahi and his co-workers, alteration on chromosome 9 is significantly related to shorter overall survival in adult B-precursor ALL when compared to patients with normal karyotypes [15]. Our findings were compatible with previous studies [15, 16], where the percentage of deletion on chromosome 9p is around 9-14%. This alteration region contains tumor suppressor genes such as CDKN2A, CDKN2B and CDK4 which might be involved in leukemogenesis [14]. CDKN2B gene plays an important role in cell cycle. Deletion of this gene may lead to loss of G1 control of cell cycle [17].
Amplification of chromosome 12q13.13 was found in 39% adult ALL samples. This alteration region contains HOTAIR, MIR196A2, MIR615, FLJ12825, LOC100240735 genes and HOXC cluster genes such as HOXC13, HOXC12, HOXC11, HOXC10, HOXC9, HOXC8, HOXC6, HOXC5 and HOXC4. In human cells, the 39 HOX genes were categorized into four clusters (HOXA, HOXB, HOXC and HOXD) and located on different chromosomes 7p15.2, 17q21.32, 12q13.13 and 2q31.1 respectively. These genes play an important role in early hematopoiesis [18] and expression of different types of HOX have been identified in early stage of the differentiation [19]. Previous studies have revealed that deregulation of HOX gene may reflect leukemic hematopoiesis [20] and some other cancers such as colorectal cancer [21], breast cancer [22], renal cancer [23] melanomas [24], squamous carcinomas [25] and lymph node metastases [26]. Translocations involving HOXC and NUP98 genes have been reported in hematological malignancies [27]. Amplifications on chromosome 12q13.13 may lead to upregulated expression of HOXC genes and hematopoiesis in adult ALL.
Amplification of chromosome 8q24.21 was observed in 51% cases. This region contains PVT1 oncogene as reported in ALL. PVT1 and murine Pvt1 is a common site of both tumorigenic translocations [28] and retroviral insertions [29]. Previous studies have revealed that murine Pvt1 locus integrated by murine leukemia virus (MLV) can induce T lymphomas in mouse and rat [30]. In normal cell, PVT1 synthesizes a variety of RNAs [31], but the RNA function is still unknown and no protein products have been found [32]. Recent studies have identified that PVT1 locus contains seven miRNAs. Overexpression of these miRNAs found in late-stage B cells when compared to immature B cells, suggests that these miRNAs induced the lymphoid development [33].
In this study, amplification of chromosome 15q11.2 occurred in 49% cases, involving the HERC2P3, GOLGA6L6, GOLGA8C, BCL8, POTEB, NF1P1, LOC646214, CXADRP2, POTEB, NF1P1, LOC727924, OR4M2, OR4N4, OR4N3P and REREP3 genes. Previous studies revealed that transcriptions of BCL8 were not found in hematopoietic tissues, whereas chromosomal translocation may cause activation of BCL8 in lymphoid tissues [34]. Translocations of BCL8 involved other sites such as 22q11, 9p13, 1p32, 7p13, 12q24 and 15q22. Expression of BCL8 genes were found in all patients with 15q11-q13 abnormalities [34]. Deletion of chromosome 16p11.2 was seen in 34% cases in this study. Recent studies indicated that deregulation of protein translation may also contribute to cancer development [33]. This alteration region contains EIF3C gene and plays an important role in translation of the initiation of protein synthesis with 13 non-identical subunits to form a multiple protein complex. Alteration of EIF3C expression may contribute to abnormal cell growth and malignant transformation.
Chromosome alterations especially translocation or inversion at chromosome 14q11.2 are frequently related with expression of T-cell receptor (TCR) gene in leukemia [35]. This chromosome region contains TCR alpha and delta genes, which encode α- and δ-chains. TCR is a unique transmembrane heterodimer composed of two different polypeptide chains [36]. Previous studies showed nonrandom breakpoints within this locus in about 30-35% of T-ALL cases. Some of the breakpoint regions have been reported, such as t(1;14)(p32;q11.2) [37], t(7;14)(p15.1;q11.2) [38], t(8;14)(q24.1;q11.2) [39], t(10;14)(q24;q11.2) [40], t(11;14)(p13;q11.2) [41], t(11;14)(p15;q11.2) [42], t(14;14)(q11.2;q32.1) [43], inv(14)(q11.2 q32.1) [43] and t(14;21)(q11.2;q22.1) [44]. In this study, we have found that some of the chromosomal alterations occurred in the 14q11.2 region, including amplifications of chromosome 14q11.2: 22,289,788-22,395,466 (46%), 14q11.2: 22,640,781 - 22,739,590 (39%), 14q11.2: 22,980,171 - 23,047,312 (17%) and deletion of chromosome 14q11.2: 22,865,854 - 22,978,881 (22%). Alterations of TCR alpha and delta genes on chromosome 14q11.2 may lead to malignant transformation of T cell progenitors, resulting from the deregulate expression of the transcription factor oncogenes controlled by TCR.
In this preliminary study, we identified 43 chromosomal alteration regions. Some of these chromosomal alteration regions are novel findings such as deletions on chromosome 1p31.1 and 3q26.1. These regions occurred frequently in adult ALL and have not been reported in previous studies. The analysis of chromosomal alterations has increased our understanding of leukemogenesis in adult ALL and has provided important information for diagnosis and prognosis in the future. Further studies are needed to characterize the cancer-related genes at the altered regions in adult ALL.
Acknowledgments
The authors thank the Director General of Health, Malaysia, for permission to publish this scientific paper. We would also like to thank Deputy Director General of Health (Research & Technical Support, Ministry of Health Malaysia) and the Director of the Institute for Medical Research for their support. This research was funded by the Ministry of Health Malaysia (JPP-IMR 09-018).
| References | ▴Top |
- Faderl S, Kantarjian HM, Talpaz M, Estrov Z. Clinical significance of cytogenetic abnormalities in adult acute lymphoblastic leukemia. Blood. 1998;91(11):3995-4019.
pubmed - Teitell MA, Pandolfi PP. Molecular genetics of acute lymphoblastic leukemia. Annu Rev Pathol. 2009;4:175-198.
doi pubmed - Devine SM, Larson RA. Acute leukemia in adults: recent developments in diagnosis and treatment. CA Cancer J Clin. 1994;44(6):326-352.
doi - Gmidene A, Sennana H, Elghezal H, Ziraoui S, Youssef YB, Elloumi M, Issaoui L, et al. Cytogenetic analysis of 298 newly diagnosed cases of acute lymphoblastic leukaemia in Tunisia. Hematol Oncol. 2008;26(2):91-97.
doi pubmed - Heinrichs S, Li C, Look AT. SNP array analysis in hematologic malignancies: avoiding false discoveries. Blood. 2010;115(21):4157-4161.
doi pubmed - Jaroslaw P. Maciejewski, Ramon V. Tiu, Christine O'Keefe. Application of array-based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br J Haematol. 2009;146:479-488.
doi pubmed - Hollox EJ, Barber JC, Brookes AJ, Armour JA. Defensins and the dynamic genome: what we can learn from structural variation at human chromosome band 8p23.1. Genome Res. 2008;18(11):1686-1697.
doi pubmed - Chng WJ, Gertz MA, Chung TH, Van Wier S, Keats JJ, Baker A, Bergsagel PL, et al. Correlation between array-comparative genomic hybridization-defined genomic gains and losses and survival: identification of 1p31-32 deletion as a prognostic factor in myeloma. Leukemia. 2010;24(4):833-842.
doi pubmed - Oshiro A, Tagawa H, Ohshima K, Karube K, Uike N, Tashiro Y, Utsunomiya A, et al. Identification of subtype-specific genomic alterations in aggressive adult T-cell leukemia/lymphoma. Blood. 2006;107(11):4500-4507.
doi pubmed - Nakashima Y, Tagawa H, Suzuki R, Karnan S, Karube K, Ohshima K, Muta K, et al. Genome-wide array-based comparative genomic hybridization of natural killer cell lymphoma/leukemia: different genomic alteration patterns of aggressive NK-cell leukemia and extranodal Nk/T-cell lymphoma, nasal type. Genes Chromosomes Cancer. 2005;44(3):247-255.
doi pubmed - Diaz MO, Rubin CM, Harden A, Ziemin S, Larson RA, Le Beau MM, Rowley JD. Deletions of interferon genes in acute lymphoblastic leukemia. N Engl J Med. 1990;322(2):77-82.
doi pubmed - Lai JL, Fenaux P, Pollet JP, Estienne MH, Savary JB, Huart JJ, Deminatti M. Acute lymphocytic leukemia with 9p anomalies. A report of four additional cases and review of the literature. Cancer Genet Cytogenet. 1988;33(1):99-109.
doi - Cayuela JM, Madani A, Sanhes L, Stern MH, Sigaux F. Multiple tumor-suppressor gene 1 inactivation is the most frequent genetic alteration in T-cell acute lymphoblastic leukemia. Blood. 1996;87(6):2180-2186.
pubmed - Kowalczyk J, Sandberg AA. A possible subgroup of ALL with 9p. Cancer Genet Cytogenet. 1983;9(4):383-385.
doi - Nahi H, Hagglund H, Ahlgren T, Bernell P, Hardling M, Karlsson K, Lazarevic V, et al. An investigation into whether deletions in 9p reflect prognosis in adult precursor B-cell acute lymphoblastic leukemia: a multi-center study of 381 patients. Haematologica. 2008;93(11):1734-1738.
doi pubmed - Matteucci C, Barba G, Varasano E, Vitale A, Mancini M, Testoni N, Cuneo A, et al. Rescue of genomic information in adult acute lymphoblastic leukaemia (ALL) with normal/failed cytogenetics: a GIMEMA centralized biological study. Br J Haematol. 2010;149(1):70-78.
doi pubmed - Hebert J, Cayuela JM, Berkeley J, Sigaux F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood. 1994;84(12):4038-4044.
pubmed - Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26(47):6766-6776.
doi pubmed - Estela Maria Novak, Eduardo Magalhaes Rego. Physiopathogenesis of Hematological Cancer. Brazil: University of Sao Paulo; 2012.
- Celetti A, Barba P, Cillo C, Rotoli B, Boncinelli E, Magli MC. Characteristic patterns of HOX gene expression in different types of human leukemia. Int J Cancer. 1993;53(2):237-244.
doi pubmed - De Vita G, Barba P, Odartchenko N, Givel JC, Freschi G, Bucciarelli G, Magli MC, et al. Expression of homeobox-containing genes in primary and metastatic colorectal cancer. Eur J Cancer. 1993;29A(6):887-893.
doi - Chariot A, Castronovo V, Le P, Gillet C, Sobel ME, Gielen J. Cloning and expression of a new HOXC6 transcript encoding a repressing protein. Biochem J. 1996;319 (Pt 1):91-97.
pubmed - Cillo C, Barba P, Freschi G, Bucciarelli G, Magli MC, Boncinelli E. HOX gene expression in normal and neoplastic human kidney. Int J Cancer. 1992;51(6):892-897.
doi pubmed - Care A, Silvani A, Meccia E, Mattia G, Stoppacciaro A, Parmiani G, Peschle C, et al. HOXB7 constitutively activates basic fibroblast growth factor in melanomas. Mol Cell Biol. 1996;16(9):4842-4851.
pubmed - Rieger E, Bijl JJ, van Oostveen JW, Soyer HP, Oudejans CB, Jiwa NM, Walboomers JM, et al. Expression of the homeobox gene HOXC4 in keratinocytes of normal skin and epithelial skin tumors is correlated with differentiation. J Invest Dermatol. 1994;103(3):341-346.
doi pubmed - Miller GJ, Miller HL, van Bokhoven A, Lambert JR, Werahera PN, Schirripa O, Lucia MS, et al. Aberrant HOXC expression accompanies the malignant phenotype in human prostate. Cancer Res. 2003;63(18):5879-5888.
pubmed - Taketani T, Taki T, Shibuya N, Kikuchi A, Hanada R, Hayashi Y. Novel NUP98-HOXC11 fusion gene resulted from a chromosomal break within exon 1 of HOXC11 in acute myeloid leukemia with t(11;12)(p15;q13). Cancer Res. 2002;62(16):4571-4574.
pubmed - Graham M, Adams JM. Chromosome 8 breakpoint far 3' of the c-myc oncogene in a Burkitt's lymphoma 2;8 variant translocation is equivalent to the murine pvt-1 locus. EMBO J. 1986;5(11):2845-2851.
pubmed - Graham M, Adams JM, Cory S. Murine T lymphomas with retroviral inserts in the chromosomal 15 locus for plasmacytoma variant translocations. Nature. 1985;314(6013):740-743.
doi pubmed - Huppi K, Siwarski D. Chimeric transcripts with an open reading frame are generated as a result of translocation to the Pvt-1 region in mouse B-cell tumors. Int J Cancer. 1994;59(6):848-851.
doi - Huppi K, Siwarski D, Skurla R, Klinman D, Mushinski JF. Pvt-1 transcripts are found in normal tissues and are altered by reciprocal(6;15) translocations in mouse plasmacytomas. Proc Natl Acad Sci U S A. 1990;87(18):6964-6968.
doi pubmed - Shtivelman E, Bishop JM. Effects of translocations on transcription from PVT. Mol Cell Biol. 1990;10(4):1835-1839.
pubmed - Huppi K, Volfovsky N, Runfola T, Jones TL, Mackiewicz M, Martin SE, Mushinski JF, et al. The identification of microRNAs in a genomically unstable region of human chromosome 8q24. Mol Cancer Res. 2008;6(2):212-221.
doi pubmed - Silvia Rasi, Gianluca Gaidano. BCL8 (B-cell CLL/lymphoma 8). Atlas Genet Cytogenet Oncol Haematol. 2008;12(6):781-784.
- Boehm T, Baer R, Lavenir I, Forster A, Waters JJ, Nacheva E, Rabbitts TH. The mechanism of chromosomal translocation t(11;14) involving the T-cell receptor C delta locus on human chromosome 14q11 and a transcribed region of chromosome 11p15. EMBO J. 1988;7(2):385-394.
pubmed - Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia. 2006;20(9):1496-1510.
doi pubmed - Huret JL. 1p32 rearrangements. Atlas Genet Cytogenet Oncol Haematol. 1998;2(1):90-92.
- Bergeron J, Macintyre E, Asnafi V. t(7;14)(p15;q11). Atlas Genet Cytogenet Oncol Haematol. 2006;10(4):279-281.
- Huret JL. t(8;21)(q22;q22) in treatment related leukemia. Atlas Genet Cytogenet Oncol Haematol. 2004;8(1):30.
- Perot C. t(10;14)(q24;q11); t(7;10)(q34;q24). Atlas Genet Cytogenet Oncol Haematol. 1999;3(3):156-157.
- Bilhou-Nabera C. t(11;14)(p13;q11); t(7;11)(q35;p13). Atlas Genet Cytogenet Oncol Haematol. 1998;3(1):22-23.
- Boyer J. t(11;14)(p15;q11). Atlas Genet Cytogenet Oncol Haematol. 2002;7(1):29-31.
- Boyer J. t(7;14)(q35;q32.1) TRB@/TCL1A; inv(14)(q11q32.1) TRA@-TRD@/TCL1A; t(14;14)(q11;q32.1) TRA@-TRD@/TCL1A. Atlas Genet Cytogenet Oncol Haematol. 2001;5(3):197-200.
- Boyer J. t(14;21)(q11;q22). Atlas Genet Cytogenet Oncol Haematol. 2004;9(1):26-27.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.